Storia
Nei primi anni del 1900, i ricercatori hanno scoperto che l’inclusione di grandi quantità di albume crudo nelle diete dei ratti produceva sintomi di tossicità. Nel 1926, Boas si riferì a questi sintomi di tossicità come sindrome da lesioni da albume. I risultati principali includevano gravi dermatiti, perdita di capelli e mancanza di coordinazione muscolare. Boas notò anche che il lievito, il fegato e diversi altri alimenti contenevano una sostanza che proteggeva i ratti dalla sindrome da lesioni da albume. Una ricerca di questo fattore protettivo ha portato alla scoperta nel 1936 della biotina.
La base biochimica per la sindrome da lesioni dell’albume è stata rapidamente chiarita quando si è scoperto che l’albume crudo contiene la glicoproteina avidina, che ha una notevole affinità per la biotina. Il legame biotina-avidina è essenzialmente irreversibile; di conseguenza, la biotina non viene liberata dal cibo e il complesso biotina-avidina viene perso nelle feci. Il passo finale per risolvere il mistero della sindrome da lesione dell’albume è stata la dimostrazione che la sindrome potrebbe essere prevenuta riscaldando l’albume, un processo che denatura l’avidina e distrugge la sua affinità per la biotina.
Struttura
La biotina è una molecola biciclica composta da un anello ureido fuso con un anello tetraidrotiofenico.
L’anello ureido è coinvolto nel legame ad alta affinità della biotina all’avidina, una glicoproteina presente nell’albume. Un sostituto dell’acido valerico è attaccato a uno dei 2 atomi di carbonio dell’anello tetraidrotiofenico. Attraverso questo gruppo carbossilico, la biotina è legata covalentemente al gruppo β-amminico della lisina in 5 carbossilasi che svolgono ruoli critici nel metabolismo intermedio.
FUNZIONI DELLA BIOTINA
Oltre al ben noto ruolo della biotina come cofattore nelle reazioni di carbossilazione, studi recenti hanno dimostrato che la biotina gioca ruoli importanti nella regolazione dell’espressione genica e della funzione immunitaria.
Reazioni di carbossilazione dipendenti dalla biotina
La biotina funziona come coenzima nelle reazioni di carbossilazione che coinvolgono il metabolismo di lipidi, glucosio e aminoacidi. Ci sono 5 carbossilasi biotina-dipendenti, ognuna delle quali esiste come apoforma inattiva. L’enzima olocarbossilasi sintetasi (HCLS ) catalizza l’aggiunta di biotina (biotinilazione) all’apoforma inattiva, che porta alla formazione della carbossilasi attiva. In tutte e 5 le carbossilasi, la biotina funziona come un coenzima o gruppo prostetico che serve come trasportatore di CO2 in una reazione multistep.
Le cinque carbossilasi biotina-dipendenti e le loro funzioni sono descritte brevemente qui sotto:
La piruvato carbossilasi (PC) catalizza la formazione di ossalacetato dal piruvato, un passo importante nel ciclo TCA, nella gluconeogenesi e nella lipogenesi; la mancanza di questa funzione può portare a ipoglicemia, chetosi e acidosi lattica.
Propionil-CoA carbossilasi (PCC) catalizza la conversione del propionil CoA in metilmalonil CoA, che a sua volta si isomerizza in succinil CoA, ed entra nel ciclo TCA (di Kreb). Il PCC è importante nel metabolismo degli acidi grassi a catena dispari e degli aminoacidi isoleucina, valina, metionina e treonina. La mancanza di questa funzione enzimatica può portare all’acidemia propionica. I livelli di PCC nei linfociti sono un indicatore sensibile dello stato della biotina.
3-Methylcrotonoyl-CoA carboxylase (MCC) è coinvolto nel catabolismo dell’aminoacido a catena ramificata, leucina. La mancanza di biotina può portare alla deviazione dei prodotti del catabolismo della leucina in una via catabolica alternativa che porta alla produzione di acido 3-idrossiisvalerico che viene poi escreto nelle urine.
L’acetil-CoA carbossilasi I (ACC I) catalizza la conversione dell’acetil CoA in malonil CoA, nel citosol, un passo importante nella sintesi dei lipidi.
L’acetil-CoA carbossilasi II (ACC II), catalizza una reazione identica nei mitocondri; il malonil CoA risultante svolge un ruolo di regolazione nell’ossidazione degli acidi grassi.
ACC I è un enzima citosolico; le rimanenti carbossilasi si trovano nei mitocondri.
Le manifestazioni cliniche della carenza di biotina possono verificarsi anche in seguito a disturbi genetici che causano una carenza dell’enzima olocarbossilasi sintetasi o carenze dei singoli enzimi carbossilasi.
Espressione genica
Gli studi hanno dimostrato che la biotinilazione degli istoni può avere un ruolo nell’espressione genica. Siti multipli che si legano alla biotina sono stati identificati negli istoni umani. La biotina può anche influenzare l’espressione genica con altri meccanismi. Alcune migliaia di geni biotina-dipendenti sono noti nelle cellule umane. Alcuni dei geni influenzati dalla biotina includono quelli che codificano per gli enzimi coinvolti nel metabolismo del glucosio (ad esempio la glucochinasi), citochine come l’interleuchina-2 e il recettore dell’insulina.
Funzione immunitaria
Gli studi suggeriscono un ruolo della biotina nella produzione di anticorpi, nella funzione dei macrofagi, nella differenziazione dei linfociti T e B e nella normale funzione delle cellule killer naturali. Le infezioni ricorrenti, specialmente quelle fungine, sono comuni nei pazienti con carenza di biotina.
Ruolo della biotina nelle condizioni neurologiche ad alta dose di biotina
La malattia dei gangli della base (BTBGD) è una rara condizione neurologica che può presentarsi con convulsioni ed encefalopatia che progredisce fino al coma e alla morte. La biotina ad alte dosi (5-10 mg/kg/giorno) è stata usata per trattare con successo questa condizione, ma il meccanismo d’azione è sconosciuto.
Recentemente, il trattamento con biotina ad alte dosi (100-300 mg/giorno) è stato trovato per migliorare i sintomi in un sottogruppo di pazienti con sclerosi multipla. Si pensa che il miglioramento dei sintomi neurologici può coinvolgere una migliore produzione di mielina secondaria all’effetto della biotina ad alte dosi sulla sintesi degli acidi grassi a catena lunga.
Ruolo della biotina in individui con disturbi di capelli, pelle e unghie
Gli integratori di biotina sono ampiamente utilizzati da coloro che sperano di ottenere capelli, pelle e unghie più sani. Tuttavia, ci sono prove limitate dell’efficacia della biotina per questo uso. La biotina in dosi elevate è stata trovata utile in due condizioni rare: la sindrome familiare dei capelli incombustibili e la sindrome delle unghie fragili. In uno studio su 541 donne che presentavano perdita di capelli, i livelli sierici di biotina sono risultati bassi nel 38% dei pazienti. L’autore ha concluso che l’eziologia della perdita di capelli è multifattoriale e gli integratori di biotina possono essere considerati se la carenza di biotina è stata dimostrata e altre cause sono state escluse.
Fonti di biotina
La biotina è presente in una grande varietà di alimenti (carni, latticini, verdure, semi e noci) ed è anche prodotta dai batteri intestinali. Inoltre, una parte sostanziale degli individui può consumare integratori alimentari contenenti biotina.
Fisiologia della biotina
La biotina ingerita è presente in forme libere e legate alle proteine. Le forme legate alle proteine sono digerite dalle proteasi gastrointestinali e dalle peptidasi per formare biocitina e biotina-oligopeptidi. La biotina libera viene rilasciata dalla biocitina e dai biotina-oligopeptidi dall’azione della biotinidasi intestinale. La biotina libera viene poi assorbita nell’intestino tenue attraverso un meccanismo mediato da un trasportatore dipendente dal Na+, che trasporta anche altri due nutrienti, l’acido pantotenico e il lipoato, ed è quindi noto come trasportatore multivitaminico dipendente dal sodio (SMVT). Il gene SMVT umano si trova sul cromosoma 2p23. L’attività del SMVT è regolata dai livelli di biotina; è up-regolata in caso di carenza di biotina e down-regolata in caso di sovralimentazione di biotina. La biotina sintetizzata battericamente è presente in forma non legata e viene assorbita nell’intestino crasso con un meccanismo simile mediato da un vettore. La produzione giornaliera combinata di biotina nelle urine e nelle feci supera l’assunzione alimentare di biotina, suggerendo l’importante ruolo svolto dalla flora intestinale come fonte di biotina.
Una volta assorbita, la biotina diventa disponibile per vari processi di biotinilazione. La biotina si lega a ciascuna delle 5 apocarbossilasi per formare l’olocarbossilasi corrispondente (vedi l’immagine qui sotto) attraverso l’azione dell’enzima olocarbossilasi sintetasi.
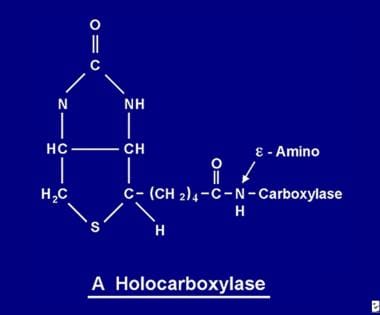 La molecola di biotina è legata alla proteina da un legame peptidico a un gruppo e-amino di un’apocarbossilasi per formare un’olocarbossilasi.
La molecola di biotina è legata alla proteina da un legame peptidico a un gruppo e-amino di un’apocarbossilasi per formare un’olocarbossilasi. Riciclaggio della biotina
Dopo che l’enzima olocarbossilasi ha effettuato diverse carbossilazioni, viene catturato dai lisosomi cellulari. Nei lisosomi, vari enzimi proteolitici degradano l’olocarbossilasi per formare biocitina, che, a sua volta, viene idrolizzata dall’enzima biotinidasi per formare biotina e lisina. La biotina libera è quindi disponibile per l’inserimento in una apocarbossilasi per formare una nuova molecola di olocarbossilasi. Questo processo di riciclaggio non è efficiente al 100%. Di conseguenza, piccole quantità di biotina libera (e un po’ di biocitina) sfuggono al ciclo e vengono perse nelle feci e nelle urine. Per questo motivo, la biotina deve essere fornita all’intestino, per reintegrare la biotina persa dal corpo. I passi coinvolti nel riciclaggio della biotina – il suo ingresso nell’intestino, il suo assorbimento, la sua incorporazione nelle olocarbossilasi, che a loro volta vengono scisse per liberare la biotina libera – costituiscono il ciclo della biotina ed è raffigurato nell’immagine qui sotto.
L’enzima biotinidasi è essenziale per il riciclaggio della biotina e gli individui con carenza di biotinidasi presenteranno quindi segni e sintomi di carenza di biotina.
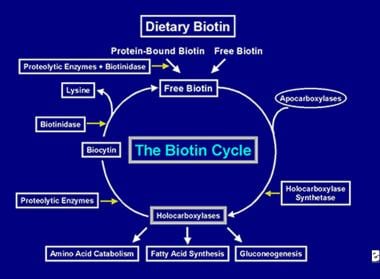 Rappresentazione del flusso di biotina nel ciclo della biotina.
Rappresentazione del flusso di biotina nel ciclo della biotina. CAUSE DI Carenza di biotina
Come già detto, la biotina è ampiamente disponibile negli alimenti, è anche prodotta dalla flora intestinale ed è ampiamente riciclata nel corpo con l’aiuto dell’enzima biotinidasi; quindi la carenza di biotina in individui sani con una dieta normale è rara. Le condizioni che possono portare a una carenza di biotina sono descritte di seguito:
Consumo eccessivo di albume crudo: L’avidina negli albumi crudi ha un’alta affinità per la biotina, rendendola indisponibile per l’assorbimento. Il calore distrugge l’avidina, quindi chi mangia uova cotte non è a rischio di carenza di biotina. Gli albumi crudi portano alla carenza di biotina solo se mangiati in quantità eccessive (forse una dozzina o più al giorno).
Nutrizione parenterale totale senza integrazione di biotina: Sono stati riportati diversi casi di carenza di biotina in pazienti che ricevono una terapia prolungata di nutrizione parenterale totale (TPN) senza aggiunta di biotina. Pertanto, tutti i pazienti che ricevono TPN devono anche ricevere biotina alla dose giornaliera raccomandata, soprattutto se si prevede che la terapia TPN duri più di 1 settimana. Tutte le farmacie ospedaliere attualmente includono la biotina nelle preparazioni TPN.
Uso di formule per bambini con biotina inadeguata: la carenza di biotina è stata segnalata in bambini che ricevono formule ipoallergeniche.
Terapia anticonvulsivante cronica: L’uso prolungato degli anticonvulsivanti, fenobarbital, fenitoina, primidone e carbamazepina, sono stati collegati alla carenza di biotina. I possibili meccanismi includono l’inibizione dell’assorbimento della biotina attraverso la mucosa intestinale, il catabolismo accelerato della biotina e l’alterato riassorbimento renale della biotina. Pertanto, la biotina supplementare è stata suggerita per i pazienti che sono trattati con anticonvulsivanti che sono stati collegati alla carenza di biotina.
Terapia antibiotica orale prolungata: L’uso prolungato di antibiotici orali è stato associato alla carenza di biotina. L’inibizione della flora intestinale che produce biotina è presumibilmente alla base della carenza di biotina. Un altro possibile meccanismo potrebbe essere la crescita eccessiva indotta dagli antibiotici di batteri che consumano biotina.
Fumo e alcolismo cronico: Gli studi hanno dimostrato che il fumo può accelerare il catabolismo della biotina, soprattutto nelle donne. L’alcolismo cronico può causare un malassorbimento intestinale della biotina.
Sindrome dell’intestino corto e malattie infiammatorie intestinali: Gli individui con sindrome dell’intestino corto e malattia infiammatoria intestinale sono anche a rischio di carenza di biotina come risultato del malassorbimento intestinale di biotina.
Carenza marginale di biotina durante la gravidanza e l’allattamento: Studi recenti hanno mostrato una diminuzione dei livelli di biotina in una percentuale significativa di donne in gravidanza e in allattamento. Si teme che una carenza marginale di biotina durante la gravidanza possa essere teratogena e alcuni esperti hanno raccomandato una maggiore assunzione di biotina da parte delle donne incinte.
Certi errori congeniti del metabolismo della biotina possono anche portare alla manifestazione della carenza di biotina.
La carenza di biotinidasi (BTD) è ereditata in modo autosomico recessivo e si verifica con una frequenza di circa 1 su 60.000 nati vivi; si stima che 1 su 120 individui sia eterozigote per questa condizione. Negli individui omozigoti per il disturbo, i livelli di biotinidasi sono < 30% normale che porta alla carenza di biotina da insufficiente rilascio di biotina libera a causa della diminuzione del riciclaggio della biotina. I sintomi del BTD si sviluppano tipicamente tra 1 settimana e 1 anno di età. La gravità della carenza enzimatica può variare. Quelli con deficit profondo di biotinidasi hanno livelli di BTD inferiori al 10% normale, mentre quelli con deficit parziale di biotinidasi hanno livelli enzimatici tra il 10-30% normale. Negli Stati Uniti e in molti altri paesi, lo screening neonatale include test per il deficit di BTD. Sono state riportate circa 150 mutazioni nel gene BTD che causano il deficit di biotinidasi. Il gene BTD si trova sul cromosoma 3p25. Un modello murino di deficit di biotinidasi è stato sviluppato per studiare vari aspetti del disturbo.
Il deficit di Holocarboxylase Synthetase (HCLS) è anche un disordine autosomico recessivo e può essere diagnosticato prenatalmente. Come discusso in precedenza, l’enzima HCLS è richiesto per la biotinilazione degli enzimi apocarbossilasi nelle forme attive dell’olocarbossilasi; quindi la carenza porta alla carenza di carbossilasi multiple. I neonati con questo disturbo si presentano nei primi mesi di vita con acidosi, iperammonemia, ipotonia, convulsioni e ritardo nello sviluppo. Le mutazioni nel gene HCLS causano il deficit di HCLS.
Perché sia il deficit di biotinidasi che quello di HCLS portano a una diminuzione dei livelli delle carbossilasi dipendenti dalla biotina, le due condizioni sono state anche classificate come deficit multiplo di carbossilasi. La carenza profonda di biotinidasi era precedentemente conosciuta come carenza di carbossilasi multipla ad esordio precoce, la carenza parziale di biotinidasi come carenza di carbossilasi multipla ad esordio tardivo o giovanile e la carenza di HCLS come carenza di carbossilasi multipla neonatale o ad esordio precoce.
Raramente, possono verificarsi anche carenze isolate di ciascuna delle cinque singole carbossilasi biotina-dipendenti.
È stata descritta anche una carenza di biotina dovuta a un difetto nel trasporto della biotina.
A prescindere dall’eziologia della carenza di biotina, le manifestazioni cliniche sono simili. Tuttavia, l’età di insorgenza, i tassi di sviluppo dei sintomi e la sequenza in cui i sintomi appaiono possono differire notevolmente. Tutti i meccanismi responsabili dello sviluppo delle manifestazioni non sono stati stabiliti.