Historia
A principios del siglo XX, los investigadores descubrieron que la inclusión de grandes cantidades de claras de huevo crudas en las dietas de las ratas producía síntomas de toxicidad. En 1926, Boas denominó a estos síntomas de toxicidad síndrome de lesión por clara de huevo. Los principales hallazgos incluían dermatitis severa, pérdida de pelo y falta de coordinación muscular. Boas también observó que la levadura, el hígado y otros alimentos contenían una sustancia que protegía a las ratas del síndrome de la clara de huevo. La búsqueda de este factor protector condujo al descubrimiento de la biotina en 1936.
La base bioquímica del síndrome de lesión de la clara de huevo se dilucidó rápidamente cuando se descubrió que las claras de huevo crudas contenían la glicoproteína avidina, que tiene una notable afinidad por la biotina. El enlace biotina-avidina es esencialmente irreversible; como resultado, la biotina no se libera de los alimentos, y el complejo biotina-avidina se pierde en las heces. El último paso para resolver el misterio del síndrome de lesión de la clara de huevo fue la demostración de que el síndrome podía prevenirse calentando las claras de huevo, un proceso que desnaturaliza la avidina y destruye su afinidad por la biotina.
Estructura
La biotina es una molécula bicíclica compuesta por un anillo ureido fusionado con un anillo de tetrahidrotiofeno.
El anillo ureido participa en la unión de alta afinidad de la biotina a la avidina, una glucoproteína que se encuentra en la clara de huevo. Un sustituto del ácido valérico está unido a uno de los 2 átomos de carbono del anillo de tetrahidrotiofeno. A través de este grupo carboxilo, la biotina se une covalentemente al grupo β-amino de la lisina en 5 carboxilasas que desempeñan papeles críticos en el metabolismo intermediario.
Funciones de la biotina
Además del conocido papel de la biotina como cofactor en las reacciones de carboxilación, estudios recientes han demostrado que la biotina desempeña papeles importantes en la regulación de la expresión genética y la función inmunitaria.
Reacciones de carboxilación dependientes de la biotina
La biotina funciona como coenzima en las reacciones de carboxilación que implican el metabolismo de los lípidos, la glucosa y los aminoácidos. Existen 5 carboxilasas dependientes de biotina, cada una de las cuales existe como una apoforma inactiva. La enzima holocarboxilasa sintetasa (HCLS ) cataliza la adición de biotina (biotinilación) a la apoforma inactiva, lo que conduce a la formación de la carboxilasa activa. En las 5 carboxilasas, la biotina funciona como coenzima o grupo prostético que sirve como portador de CO2 en una reacción de varios pasos.
A continuación se describen brevemente las cinco carboxilasas dependientes de biotina y sus funciones:
La piruvato carboxilasa (PC) cataliza la formación de oxaloacetato a partir del piruvato, un paso importante en el ciclo del TCA, la gluconeogénesis y la lipogénesis; la falta de esta función puede provocar hipoglucemia, cetosis y acidosis láctica.
La propionil-CoA carboxilasa (PCC) cataliza la conversión de propionil-CoA en metilmalonil-CoA, que a su vez se isomeriza en succinil-CoA, y entra en el ciclo TCA (de Kreb). La PCC es importante en el metabolismo de los ácidos grasos de cadena extraña y de los aminoácidos isoleucina, valina, metionina y treonina. La falta de esta función enzimática puede provocar acidemia propiónica. Los niveles de PCC en los linfocitos son un indicador sensible del estado de la biotina.
La 3-metilcrotonoil-CoA carboxilasa (MCC) participa en el catabolismo del aminoácido de cadena ramificada, la leucina. La falta de biotina puede conducir a la derivación de los productos del catabolismo de la leucina a una vía catabólica alternativa que conduce a la producción de ácido 3-hidroxisovalérico que se excreta en la orina.
La acetil-CoA carboxilasa I (ACC I) cataliza la conversión de acetil CoA en malonil CoA, en el citosol, un paso importante en la síntesis de lípidos.
La acetil-CoA carboxilasa II (ACC II), cataliza una reacción idéntica en la mitocondria; la malonil CoA resultante desempeña un papel regulador en la oxidación de los ácidos grasos.
La ACC I es una enzima citosólica; las restantes carboxilasas se encuentran en la mitocondria.
Las manifestaciones clínicas de la deficiencia de biotina también pueden producirse como resultado de trastornos genéticos que causan una deficiencia de la enzima holocarboxilasa sintetasa o deficiencias de las enzimas carboxilasas individuales.
Expresión génica
Los estudios han demostrado que la biotinilación de las histonas puede desempeñar un papel en la expresión génica. Se han identificado múltiples sitios que se unen a la biotina en las histonas humanas. La biotina también puede afectar a la expresión génica por otros mecanismos. Se conocen unos cuantos miles de genes dependientes de la biotina en las células humanas. Algunos de los genes en los que influye la biotina son los que codifican las enzimas que intervienen en el metabolismo de la glucosa (por ejemplo, la glucoquinasa), las citoquinas como la interleucina-2 y el receptor de la insulina.
Función inmunitaria
Los estudios sugieren un papel de la biotina en la producción de anticuerpos, la función de los macrófagos, la diferenciación de los linfocitos T y B, así como la función normal de las células asesinas naturales. Las infecciones recurrentes, especialmente por hongos, son comunes en pacientes con deficiencia de biotina.
Función de la biotina en las condiciones neurológicas que responden a altas dosis de biotina
La enfermedad de los ganglios basales que responde a la biotina (BTBGD) es una condición neurológica rara que puede presentarse con convulsiones y encefalopatía que progresa hasta el coma y la muerte. Se han utilizado altas dosis de biotina ( 5-10 mg/kg/día) para tratar con éxito esta enfermedad, pero se desconoce el mecanismo de acción.
Recientemente, se ha descubierto que el tratamiento con altas dosis de biotina (100-300 mg/día) mejora los síntomas en un subconjunto de pacientes con esclerosis múltiple. Se cree que la mejora de los síntomas neurológicos puede implicar una mejora de la producción de mielina secundaria al efecto de las altas dosis de biotina en la síntesis de ácidos grasos de cadena larga.
Función de la biotina en individuos con trastornos del cabello, la piel y las uñas
Los suplementos de biotina son muy utilizados por quienes esperan conseguir un cabello, una piel y unas uñas más saludables. Sin embargo, las pruebas de la eficacia de la biotina para este uso son limitadas. Se ha comprobado que la biotina en dosis elevadas es útil en dos afecciones poco frecuentes: el síndrome del cabello familiar incombustible y el síndrome de las uñas quebradizas. En un estudio de 541 mujeres que presentaban pérdida de cabello, se comprobó que los niveles séricos de biotina eran bajos en el 38% de las pacientes. El autor concluyó que la etiología de la caída del cabello es multifactorial y que se puede considerar el uso de suplementos de biotina si se ha demostrado una deficiencia de biotina y se han descartado otras causas.
Fuentes de biotina
La biotina está presente en una amplia variedad de alimentos (carnes, lácteos, verduras, semillas y frutos secos) y también es producida por las bacterias intestinales. Además, una proporción considerable de individuos puede consumir suplementos dietéticos que contienen biotina.
Fisiología de la biotina
La biotina ingerida está presente en forma libre y unida a proteínas. Las formas unidas a proteínas son digeridas por las proteasas y peptidasas gastrointestinales para formar biocitina y biotina-oligopéptidos. La biotina libre se libera de la biocitina y de los biotina-oligopéptidos por la acción de la biotinidasa intestinal. La biotina libre se absorbe entonces en el intestino delgado a través de un mecanismo mediado por un transportador dependiente del Na+, que también transporta otros dos nutrientes, el ácido pantoténico y el lipoato, por lo que se conoce como transportador multivitamínico dependiente del sodio (SMVT). El gen humano del SMVT está localizado en el cromosoma 2p23. La actividad del SMVT está regulada por los niveles de biotina; se regula al alza en caso de deficiencia de biotina y a la baja en caso de suplemento excesivo de biotina. La biotina sintetizada bacterialmente está presente en forma no ligada y se absorbe en el intestino grueso por un mecanismo similar mediado por un transportador. La producción diaria combinada de biotina en la orina y las heces supera la ingesta dietética de biotina, lo que sugiere el importante papel que desempeña la flora intestinal como fuente de biotina.
Una vez absorbida, la biotina queda disponible para varios procesos de biotinilación. La biotina se une a cada una de las 5 apocarboxilasas para formar la correspondiente holocarboxilasa (véase la imagen inferior) mediante la acción de la enzima holocarboxilasa sintetasa.
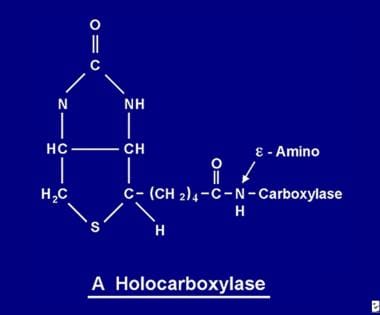 La molécula de biotina se une a la proteína mediante un enlace peptídico a un grupo e-amino de una apocarboxilasa para formar una holocarboxilasa.
La molécula de biotina se une a la proteína mediante un enlace peptídico a un grupo e-amino de una apocarboxilasa para formar una holocarboxilasa. Reciclaje de la biotina
Después de que la enzima holocarboxilasa haya realizado varias carboxilaciones es capturada por los lisosomas celulares. En los lisosomas, varias enzimas proteolíticas degradan la holocarboxilasa para formar biocitina, que, a su vez, es hidrolizada por la enzima biotinidasa para formar biotina y lisina. La biotina libre está entonces disponible para su inserción en una apocarboxilasa para formar una nueva molécula de holocarboxilasa. Este proceso de reciclaje no es 100% eficiente. Como resultado, pequeñas cantidades de biotina libre (y algo de biocitina) escapan del ciclo y se pierden en las heces y la orina. Por esta razón, es necesario suministrar biotina al intestino, para reponer la biotina perdida por el organismo. Los pasos implicados en el reciclaje de la biotina -su entrada en el intestino, su absorción, su incorporación a las holocarboxilasas, que a su vez se descomponen para liberar biotina libre- constituyen el ciclo de la biotina y se representa en la imagen siguiente.
La enzima biotinidasa es esencial para el reciclaje de la biotina y los individuos con deficiencia de biotinidasa presentarán, por tanto, signos y síntomas de deficiencia de biotina.
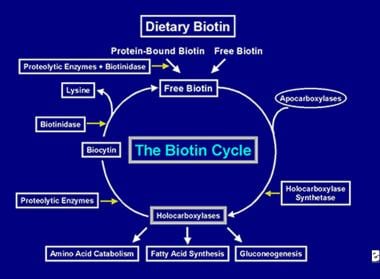 Representación del flujo de biotina en el ciclo de la biotina.
Representación del flujo de biotina en el ciclo de la biotina. Causas de la deficiencia de biotina
Como se ha mencionado anteriormente, la biotina está ampliamente disponible en los alimentos, también es producida por la flora intestinal y se recicla ampliamente en el cuerpo con la ayuda de la enzima biotinidasa; por lo tanto, la deficiencia de biotina en individuos sanos que comen una dieta normal es rara. A continuación se describen las condiciones que pueden provocar una deficiencia de biotina:
Consumo excesivo de claras de huevo crudas: La avidina de las claras de huevo crudas tiene una gran afinidad por la biotina, lo que hace que no esté disponible para su absorción. El calor destruye la avidina, por lo que quienes comen huevos cocidos no corren el riesgo de sufrir una deficiencia de biotina. Las claras de huevo crudas sólo provocan una deficiencia de biotina cuando se consumen en cantidades excesivas (quizás una docena o más al día).
Nutrición parenteral total sin suplemento de biotina: Se han notificado varios casos de deficiencia de biotina en pacientes que reciben una terapia prolongada de nutrición parenteral total (NPT) sin biotina añadida. Por lo tanto, todos los pacientes que reciben NPT deben recibir también biotina en la dosis diaria recomendada, especialmente si se espera que la terapia de NPT dure más de 1 semana. Actualmente, todas las farmacias de los hospitales incluyen biotina en los preparados de NPT.
Uso de fórmulas para lactantes con biotina inadecuada: Se ha notificado una deficiencia de biotina en lactantes que reciben fórmulas hipoalergénicas.
Terapia anticonvulsiva crónica: El uso prolongado de los anticonvulsivos, fenobarbital, fenitoína, primidona y carbamazepina, se ha relacionado con la deficiencia de biotina. Los posibles mecanismos incluyen la inhibición de la captación de biotina a través de la mucosa intestinal, la aceleración del catabolismo de la biotina y el deterioro de la reabsorción renal de la biotina. Por lo tanto, se ha sugerido un suplemento de biotina para los pacientes que son tratados con anticonvulsivos que se han relacionado con la deficiencia de biotina.
Terapia antibiótica oral prolongada: El uso prolongado de antibióticos orales se ha asociado con la deficiencia de biotina. Se presume que la inhibición de la flora intestinal que produce biotina es la base de la deficiencia de biotina. Otro posible mecanismo podría ser el sobrecrecimiento inducido por los antibióticos de las bacterias que consumen biotina.
Tabaco y alcoholismo crónico: Los estudios han demostrado que el tabaquismo puede acelerar el catabolismo de la biotina, especialmente en las mujeres. El alcoholismo crónico puede causar malabsorción intestinal de biotina.
Síndrome del intestino corto y enfermedad inflamatoria intestinal: Los individuos con síndrome de intestino corto y enfermedad inflamatoria intestinal también corren el riesgo de sufrir una deficiencia de biotina como resultado de la malabsorción intestinal de biotina.
Deficiencia marginal de biotina durante el embarazo y la lactancia: Estudios recientes han mostrado una disminución de los niveles de biotina en una proporción significativa de mujeres embarazadas y lactantes. Existe la preocupación de que una deficiencia marginal de biotina durante el embarazo pueda ser teratogénica y algunos expertos han recomendado una mayor ingesta de biotina por parte de las mujeres embarazadas.
Ciertos errores congénitos del metabolismo de la biotina también pueden dar lugar a la manifestación de una deficiencia de biotina.
La deficiencia de biotinidasa (BTD) se hereda de forma autosómica recesiva y se produce con una frecuencia aproximada de 1 de cada 60.000 nacidos vivos; se estima que 1 de cada 120 individuos son heterocigotos para la enfermedad. En los individuos homocigotos para el trastorno, los niveles de biotinidasa son < 30% normales, lo que conduce a una deficiencia de biotina por la insuficiente liberación de biotina libre debido a la disminución del reciclaje de biotina. Los síntomas de la EAB suelen aparecer entre una semana y un año de edad. La gravedad de la deficiencia enzimática puede variar. Las personas con una deficiencia profunda de biotinidasa tienen niveles de BTD inferiores al 10% de lo normal, mientras que las que tienen una deficiencia parcial de biotinidasa tienen niveles de la enzima entre el 10 y el 30% de lo normal. En EE.UU. y en muchos otros países, el cribado de los recién nacidos incluye pruebas para detectar la deficiencia de BTD. Se han notificado aproximadamente 150 mutaciones en el gen BTD que causan la deficiencia de biotinidasa. El gen BTD está localizado en el cromosoma 3p25. Se ha desarrollado un modelo de ratón de deficiencia de biotinidasa para estudiar varios aspectos del trastorno.
La deficiencia de holocarboxilasa (HCLS) es también un trastorno autosómico recesivo y puede diagnosticarse prenatalmente. Como se ha comentado anteriormente, la enzima HCLS es necesaria para la biotinilación de las enzimas apocarboxilasas en las formas holocarboxilasas activas; por lo tanto, su deficiencia conduce a una deficiencia múltiple de carboxilasas. Los bebés con este trastorno presentan en los primeros meses de vida acidosis, hiperamonemia, hipotonía, convulsiones y retraso en el desarrollo. Las mutaciones en el gen HCLS causan la deficiencia de HCLS.
Debido a que tanto la deficiencia de biotinidasa como la de HCLS conducen a una disminución de los niveles de las carboxilasas dependientes de biotina, las dos afecciones se han clasificado también como deficiencia múltiple de carboxilasas. La deficiencia profunda de biotinidasa se conocía anteriormente como deficiencia de carboxilasa múltiple de inicio temprano, la deficiencia parcial de biotinidasa como deficiencia de carboxilasa múltiple de inicio tardío o juvenil y la deficiencia de HCLS como deficiencia de carboxilasa múltiple neonatal o de inicio temprano.
Raramente, también pueden producirse deficiencias aisladas de cada una de las cinco carboxilasas individuales dependientes de biotina.
También se ha descrito la deficiencia de biotina debida a un defecto en el transporte de biotina.
Independientemente de la etiología de la deficiencia de biotina, las manifestaciones clínicas son similares. Sin embargo, la edad de inicio, las tasas de desarrollo de los síntomas y la secuencia de aparición de los mismos pueden ser muy diferentes. No se han establecido todos los mecanismos responsables del desarrollo de las manifestaciones.