Histoire
Au début des années 1900, des chercheurs ont constaté que l’inclusion de grandes quantités de blancs d’œufs crus dans les régimes alimentaires des rats produisait des symptômes de toxicité. En 1926, Boas a appelé ces symptômes de toxicité le syndrome de blessure au blanc d’œuf. Les principales constatations sont les suivantes : dermatite grave, perte de cheveux et manque de coordination musculaire. Boas a également noté que la levure, le foie et plusieurs autres aliments contenaient une substance qui protégeait les rats du syndrome de la lésion du blanc d’œuf. La recherche de ce facteur de protection a conduit à la découverte en 1936 de la biotine.
La base biochimique du syndrome de blessure du blanc d’œuf a été rapidement élucidée lorsqu’on a découvert que le blanc d’œuf cru contenait la glycoprotéine avidine, qui a une affinité remarquable pour la biotine. La liaison biotine-avidine est essentiellement irréversible ; par conséquent, la biotine n’est pas libérée des aliments et le complexe biotine-avidine est perdu dans les fèces. L’étape finale dans la résolution du mystère du syndrome de blessure au blanc d’œuf a été la démonstration que le syndrome pouvait être évité en chauffant les blancs d’œuf, un processus qui dénature l’avidine et détruit son affinité pour la biotine.
Structure
La biotine est une molécule bicyclique composée d’un cycle uréido fusionné avec un cycle tétrahydrothiophène.
Le cycle uréido est impliqué dans la liaison de haute affinité de la biotine à l’avidine, une glycoprotéine présente dans le blanc d’œuf. Un substituant acide valérique est attaché à l’un des 2 atomes de carbone du cycle tétrahydrothiophène. Grâce à ce groupe carboxyle, la biotine est liée de manière covalente au groupe β-amino de la lysine dans 5 carboxylases qui jouent des rôles critiques dans le métabolisme intermédiaire.
FONCTIONS DE LA BIOTINE
En plus du rôle bien connu de la biotine comme cofacteur dans les réactions de carboxylation, des études récentes ont montré que la biotine joue des rôles importants dans la régulation de l’expression des gènes et de la fonction immunitaire.
Réactions de carboxylation dépendantes de la biotine
La biotine fonctionne comme coenzyme dans les réactions de carboxylation impliquant le métabolisme des lipides, du glucose et des acides aminés. Il existe 5 carboxylases dépendantes de la biotine dont chacune existe sous forme d’apoforme inactive. L’enzyme holocarboxylase synthétase (HCLS ) catalyse l’addition de biotine (biotinylation) à l’apoforme inactive, ce qui conduit à la formation de la carboxylase active. Dans les 5 carboxylases, la biotine fonctionne comme un coenzyme ou un groupe prosthétique qui sert de transporteur pour le CO2 dans une réaction à plusieurs étapes.
Les cinq carboxylases dépendantes de la biotine et leurs fonctions sont décrites brièvement ci-dessous :
La pyruvate carboxylase (PC) catalyse la formation d’oxaloacétate à partir du pyruvate, une étape importante dans le cycle TCA, la gluconéogenèse et la lipogenèse ; l’absence de cette fonction peut entraîner une hypoglycémie, une cétose et une acidose lactique.
La propionyl-CoA carboxylase (PCC) catalyse la conversion du propionyl CoA en méthylmalonyl CoA, qui à son tour s’isomérise en succinyl CoA, et entre dans le cycle TCA (cycle de Kreb). La PCC est importante dans le métabolisme des acides gras à chaîne impaire et des acides aminés isoleucine, valine, méthionine et thréonine. L’absence de cette fonction enzymatique peut entraîner une acidémie propionique. Les niveaux de PCC dans les lymphocytes est un indicateur sensible du statut en biotine.
La 3-Méthylcrotonoyl-CoA carboxylase (MCC) est impliquée dans le catabolisme de l’acide aminé à chaîne ramifiée, la leucine. Un manque de biotine peut conduire à un shuntage des produits du catabolisme de la leucine dans une voie catabolique alternative conduisant à la production d’acide 3-hydoxyisovalérique qui est ensuite excrété dans l’urine.
L’acétyl-CoA carboxylase I (ACC I) catalyse la conversion de l’acétyl CoA en malonyl CoA, dans le cytosol, une étape importante dans la synthèse des lipides.
L’acétyl-CoA carboxylase II (ACC II), catalyse une réaction identique dans les mitochondries ; le malonyl CoA résultant joue un rôle régulateur dans l’oxydation des acides gras.
L’ACC I est une enzyme cytosolique ; les autres carboxylases se trouvent dans les mitochondries.
Les manifestations cliniques de la carence en biotine peuvent également survenir à la suite de troubles génétiques provoquant un déficit de l’enzyme holocarboxylase synthétase ou des déficiences des différentes enzymes carboxylases.
Expression génétique
Des études ont montré que la biotinylation des histones peut jouer un rôle dans l’expression génétique. De multiples sites qui se lient à la biotine ont été identifiés dans les histones humaines. La biotine peut également affecter l’expression des gènes par d’autres mécanismes. Quelques milliers de gènes dépendant de la biotine sont connus dans les cellules humaines. Parmi les gènes influencés par la biotine figurent ceux qui codent pour les enzymes impliquées dans le métabolisme du glucose (par exemple la glucokinase), les cytokines comme l’interleukine-2 et le récepteur de l’insuline.
Fonction immunitaire
Des études suggèrent un rôle de la biotine dans la production d’anticorps, la fonction des macrophages, la différenciation des lymphocytes T et B, ainsi que la fonction normale des cellules tueuses naturelles. Les infections récurrentes, notamment fongiques, sont fréquentes chez les patients présentant une carence en biotine.
Rôle de la biotine dans les affections neurologiques répondant à de fortes doses de biotine
La maladie des ganglions de la base répondant à la biotine-thiamine (BTBGD) est une affection neurologique rare qui peut se manifester par des crises et une encéphalopathie évoluant vers le coma et la mort. De fortes doses de biotine ( 5-10 mg/kg/jour) ont été utilisées pour traiter avec succès cette affection, mais le mécanisme d’action est inconnu.
Récemment, on a constaté qu’un traitement à forte dose de biotine (100-300 mg/jour) améliorait les symptômes chez un sous-ensemble de patients atteints de sclérose en plaques. On pense que l’amélioration des symptômes neurologiques peut impliquer une meilleure production de myéline secondaire à l’effet de la biotine à haute dose sur la synthèse des acides gras à longue chaîne.
Rôle de la biotine chez les personnes souffrant de troubles des cheveux, de la peau et des ongles
Les suppléments de biotine sont largement utilisés par ceux qui espèrent obtenir des cheveux, une peau et des ongles plus sains. Cependant, il existe des preuves limitées de l’efficacité de la biotine pour cet usage. La biotine à fortes doses s’est avérée utile dans deux pathologies rares : le syndrome familial des cheveux incomblables et le syndrome des ongles cassants. Dans une étude portant sur 541 femmes présentant une perte de cheveux, on a constaté que les taux sériques de biotine étaient faibles chez 38 % des patientes. L’auteur a conclu que l’étiologie de la perte de cheveux est multifactorielle et que des suppléments de biotine peuvent être envisagés si une carence en biotine a été démontrée et que d’autres causes ont été écartées.
Sources de biotine
La biotine est présente dans une grande variété d’aliments (viandes, produits laitiers, légumes, graines et noix) et est également produite par les bactéries intestinales. En outre, une proportion importante d’individus peut consommer des compléments alimentaires contenant de la biotine.
Physiologie de la biotine
La biotine ingérée est présente sous des formes libres et liées aux protéines. Les formes liées aux protéines sont digérées par les protéases et les peptidases gastro-intestinales pour former la biocytine et les biotine-oligopeptides. La biotine libre est libérée de la biocytine et des biotine-oligopeptides par l’action de la biotinidase intestinale. La biotine libre est ensuite absorbée dans l’intestin grêle par un mécanisme dépendant du Na+ et médié par un transporteur, qui transporte également deux autres nutriments, l’acide pantothénique et le lipoate, d’où son nom de transporteur multivitaminé dépendant du sodium (SMVT). Le gène SMVT humain est situé sur le chromosome 2p23. L’activité du SMVT est régulée par les niveaux de biotine ; elle est régulée à la hausse en cas de carence en biotine et régulée à la baisse en cas de suralimentation en biotine. La biotine synthétisée par voie bactérienne est présente sous forme non liée et est absorbée dans le gros intestin par un mécanisme similaire médié par un transporteur. La production quotidienne combinée de biotine dans l’urine et les selles dépasse l’apport alimentaire en biotine, ce qui suggère le rôle important joué par la flore intestinale comme source de biotine.
Une fois absorbée, la biotine devient disponible pour divers processus de biotinylation. La biotine se lie à chacune des 5 apocarboxylases pour former l’holocarboxylase correspondante (voir l’image ci-dessous) via l’action de l’enzyme holocarboxylase synthétase.
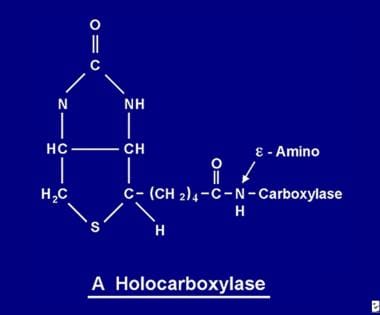 La molécule de biotine est liée à la protéine par une liaison peptidique à un groupe e-amino d’une apocarboxylase pour former une holocarboxylase.
La molécule de biotine est liée à la protéine par une liaison peptidique à un groupe e-amino d’une apocarboxylase pour former une holocarboxylase. Recyclage de la biotine
Après que l’enzyme holocarboxylase ait effectué plusieurs carboxylations, elle est capturée par les lysosomes cellulaires. Dans les lysosomes, diverses enzymes protéolytiques dégradent l’holocarboxylase pour former de la biocytine, qui, à son tour, est hydrolysée par l’enzyme biotinidase pour former de la biotine et de la lysine. La biotine libre peut alors être insérée dans une apocarboxylase pour former une nouvelle molécule d’holocarboxylase. Ce processus de recyclage n’est pas efficace à 100 %. Par conséquent, de petites quantités de biotine libre (et un peu de biocytine) échappent au cycle et sont perdues dans les fèces et l’urine. C’est pourquoi la biotine doit être fournie à l’intestin, afin de reconstituer la biotine perdue par l’organisme. Les étapes du recyclage de la biotine- son entrée dans l’intestin, son absorption, son incorporation dans les holocarboxylases, qui sont à leur tour décomposées pour libérer la biotine libre constituent le cycle de la biotine et sont représentées dans l’image ci-dessous.
L’enzyme biotinidase est essentielle au recyclage de la biotine et les individus présentant un déficit en biotinidase présenteront donc des signes et des symptômes de carence en biotine.
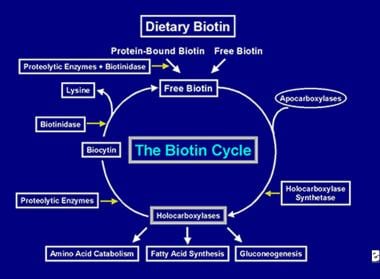 Représentation du flux de biotine dans le cycle de la biotine.
Représentation du flux de biotine dans le cycle de la biotine. CAUSES DE LA CARENCE EN BIOTINE
Comme mentionné précédemment, la biotine est largement disponible dans les aliments, est également produite par la flore intestinale et est largement recyclée dans l’organisme à l’aide de l’enzyme biotinidase ; par conséquent, la carence en biotine chez les individus en bonne santé ayant une alimentation normale est rare. Les conditions qui peuvent entraîner une carence en biotine sont décrites ci-dessous :
Consommation excessive de blancs d’œufs crus : L’avidine présente dans les blancs d’œufs crus a une forte affinité pour la biotine, ce qui la rend indisponible pour l’absorption. La chaleur détruit l’avidine, de sorte que ceux qui mangent des œufs cuits ne risquent pas d’avoir une carence en biotine. Les blancs d’œufs crus n’entraînent une carence en biotine que lorsqu’ils sont consommés en quantités excessives (peut-être une douzaine ou plus par jour).
Nutrition parentérale totale sans supplémentation en biotine : Plusieurs cas de carence en biotine chez des patients recevant une thérapie de nutrition parentérale totale (TPN) prolongée sans ajout de biotine ont été rapportés. Par conséquent, tous les patients recevant une NPT doivent également recevoir de la biotine à la dose quotidienne recommandée, en particulier si la thérapie NPT doit durer plus d’une semaine. Toutes les pharmacies hospitalières incluent actuellement de la biotine dans les préparations de NPT.
Utilisation de préparations pour nourrissons dont la biotine est insuffisante : une carence en biotine a été signalée chez des nourrissons recevant des préparations hypoallergéniques.
Traitement anticonvulsivant chronique : L’utilisation prolongée des anticonvulsivants, phénobarbital, phénytoïne, primidone et carbamazépine, a été liée à une carence en biotine. Les mécanismes possibles comprennent l’inhibition de l’absorption de la biotine par la muqueuse intestinale, l’accélération du catabolisme de la biotine et l’altération de la réabsorption rénale de la biotine. Par conséquent, une supplémentation en biotine a été suggérée pour les patients traités par des anticonvulsivants qui ont été liés à une carence en biotine.
Antibiothérapie orale prolongée : L’utilisation prolongée d’antibiotiques par voie orale a été associée à une carence en biotine. L’inhibition de la flore intestinale qui produit la biotine est présumée être à la base de la carence en biotine. Un autre mécanisme possible pourrait être la surcroissance induite par les antibiotiques des bactéries qui consomment la biotine.
Tabagisme et alcoolisme chronique : Des études ont montré que le tabagisme peut accélérer le catabolisme de la biotine, en particulier chez les femmes. L’alcoolisme chronique peut provoquer une malabsorption intestinale de la biotine.
Syndrome de l’intestin court et maladie inflammatoire de l’intestin : Les personnes atteintes du syndrome de l’intestin court et des maladies inflammatoires de l’intestin sont également exposées à un risque de carence en biotine en raison de la malabsorption intestinale de la biotine.
Déficience marginale en biotine pendant la grossesse et la lactation : Des études récentes ont montré une diminution des niveaux de biotine chez une proportion significative de femmes enceintes et allaitantes. On craint qu’une carence marginale en biotine pendant la grossesse puisse être tératogène et certains experts ont recommandé un apport plus élevé en biotine chez les femmes enceintes.
Certaines erreurs innées du métabolisme de la biotine peuvent également conduire à la manifestation d’une carence en biotine.
La déficience en biotinidase (BTD) est héritée sur un mode autosomique récessif et se produit à une fréquence d’environ 1 sur 60 000 naissances vivantes ; on estime que 1 individu sur 120 est hétérozygote pour cette affection. Chez les personnes homozygotes, le taux de biotinidase est < 30% normal, ce qui entraîne une carence en biotine due à une libération insuffisante de biotine libre en raison d’une diminution du recyclage de la biotine. Les symptômes de la BTD se développent généralement entre l’âge d’une semaine et d’un an. La gravité de la déficience enzymatique peut varier. Les personnes présentant un déficit profond en biotinidase ont des taux de BTD inférieurs à 10 % de la normale, tandis que celles présentant un déficit partiel en biotinidase ont des taux d’enzyme compris entre 10 et 30 % de la normale. Aux États-Unis et dans de nombreux autres pays, le dépistage des nouveau-nés comprend des tests de détection du déficit en BTD. Environ 150 mutations du gène BTD ont été signalées comme pouvant provoquer un déficit en biotinidase. Le gène BTD est situé sur le chromosome 3p25. Un modèle murin de déficience en biotinidase a été développé pour étudier divers aspects de ce trouble.
La déficience en holocarboxylase synthétase (HCLS) est également un trouble autosomique récessif et peut être diagnostiquée avant la naissance. Comme nous l’avons vu précédemment, l’enzyme HCLS est nécessaire à la biotinylation des enzymes apocarboxylases en formes holocarboxylases actives ; par conséquent, une déficience entraîne une déficience en carboxylases multiples. Les nourrissons atteints de cette maladie présentent au cours des premiers mois de leur vie une acidose, une hyperammonémie, une hypotonie, des crises et un retard de développement. Les mutations du gène HCLS provoquent le déficit en HCLS.
Parce que les déficits en biotinidase et en HCLS entraînent tous deux une diminution des taux de carboxylases dépendantes de la biotine, ces deux affections ont également été classées comme des déficits multiples en carboxylases. Le déficit profond en biotinidase était auparavant connu sous le nom de déficit multiple en carboxylase à début précoce, le déficit partiel en biotinidase sous le nom de déficit multiple en carboxylase à début tardif ou juvénile et le déficit en HCLS sous le nom de déficit multiple en carboxylase néonatal ou à début précoce.
Rares sont les déficits isolés de chacune des cinq carboxylases individuelles dépendantes de la biotine qui peuvent également se produire.
Un déficit en biotine dû à un défaut de transport de la biotine a également été décrit.
Qu’importe l’étiologie de la carence en biotine, les manifestations cliniques sont similaires. Cependant, l’âge d’apparition, le rythme de développement des symptômes et la séquence d’apparition des symptômes peuvent être très différents. Tous les mécanismes responsables du développement des manifestations n’ont pas été établis.