History
No início dos anos 1900, os pesquisadores descobriram que a inclusão de grandes quantidades de claras de ovo cru em dietas em ratos produzia sintomas de toxicidade. Em 1926, Boas referiu-se a esses sintomas de toxicidade como síndrome da lesão da clara do ovo. As principais descobertas incluíram dermatites graves, perda de cabelo e falta de coordenação muscular. Boas também notou que levedura, fígado e vários outros alimentos continham uma substância que protegia ratos da síndrome da lesão da clara do ovo. Uma busca por este fator de proteção levou à descoberta em 1936 da biotina.
A base bioquímica para a síndrome da lesão da clara do ovo foi rapidamente elucidada quando se descobriu que as claras de ovo cru continham a glicoproteína avidina, que tem uma afinidade notável para a biotina. A ligação biotina-avidina é essencialmente irreversível; como resultado, a biotina não é liberada dos alimentos, e o complexo biotina-avidina é perdido nas fezes. O último passo para resolver o mistério da síndrome da clara do ovo foi a demonstração de que a síndrome poderia ser evitada através do aquecimento das claras de ovo, um processo que desnaturaliza a avidina e destrói sua afinidade com a biotina.
Estrutura
Biotina é uma molécula bicíclica composta por um anel de ureido fundido com um anel de tetrahidrotiofeno.
O anel de ureido está envolvido na ligação de alta afinidade da biotina com a avidina, uma glicoproteína encontrada na clara do ovo. Um substituto do ácido valérico é ligado a um dos 2 átomos de carbono do anel de tetrahidrotiofeno. Através deste grupo carboxil, a biotina está ligada covalentemente ao grupo β-amino da lisina em 5 carboxilases que desempenham papéis críticos no metabolismo intermediário.
FUNÇÕES DA BIOTINA
Além do conhecido papel da biotina como co-fator nas reações de carboxilação, estudos recentes mostraram que a biotina desempenha papéis importantes na regulação da expressão gênica e função imune.
Reações de carboxilação dependentes da biotina
A biotina funciona como coenzima nas reações de carboxilação envolvendo o metabolismo de lipídios, glicose e aminoácidos. Existem 5 carboxilases biotínicas dependentes de carboxilases, cada uma das quais existe como um apoforma inativo. A enzima holocarboxilase sintetase (HCLS ) catalisa a adição de biotina (biotinilação) à apoforma inativa, o que leva à formação da carboxilase ativa. Em todas as 5 carboxilases, a biotina funciona como uma coenzima ou grupo protético que serve como portador de CO2 em uma reação em múltiplas etapas.
As cinco carboxilases dependentes de biotina e suas funções são descritas brevemente abaixo:
A carboxilase piruvada (PC) catalisa a formação de oxaloacetato a partir do piruvato, um passo importante no ciclo do TCA, gluconeogênese e lipogênese; a falta desta função pode levar à hipoglicemia, cetose e acidose láctica.
Propionil-CoA carboxilase (PCC) catalisa a conversão de propionil-CoA em metilmalonil-CoA, que por sua vez isomeriza a succinil-CoA, e entra no ciclo de TCA (Kreb’s). O PCC é importante no metabolismo dos ácidos gordos de cadeia ímpar, e os aminoácidos isoleucina, valina, metionina, e treonina. A falta desta função enzimática pode levar à acidemia propiônica. Os níveis de PCC nos linfócitos é um indicador sensível do estado de biotina.
3-Metilcrotonoil-CoA carboxilase (MCC) está envolvida no catabolismo do aminoácido de cadeia ramificada, a leucina. A falta de biotina pode levar ao desvio dos produtos do catabolismo leucino para um caminho catabólico alternativo que leva à produção de ácido 3-hidroxi-isovalérico que é então excretado na urina.
Acetil-CoA carboxilase I (ACC I) catalisa a conversão de acetil-CoA em malonil-CoA, no citosol, um passo importante na síntese de lipídios.
Acetil-CoA carboxilase II (ACC II), catalisa uma reacção idêntica nas mitocôndrias; a malonil-CoA resultante desempenha um papel regulador na oxidação dos ácidos gordos.
ACC I é uma enzima citosólica; as carboxilases restantes são encontradas nas mitocôndrias.
As manifestações clínicas de deficiência de biotina também podem ocorrer como resultado de desordens genéticas causando uma deficiência da enzima holocarboxilase sintetase ou deficiências das enzimas carboxilase individuais.
Expressão do gene
Estudos mostraram que a biotinilação das histonas pode ter um papel na expressão gênica. Múltiplos locais que se ligam à biotina foram identificados nas histonas humanas. A biotina também pode afetar a expressão gênica por outros mecanismos. Alguns milhares de genes dependentes da biotina são conhecidos em células humanas. Alguns dos genes influenciados pela biotina incluem aqueles que codificam as enzimas envolvidas no metabolismo da glicose (por exemplo, glucocinase), citocinas como a interleucina-2 e o receptor de insulina.
Função imune
Estudos sugerem um papel da biotina na produção de anticorpos, função macrofágica, diferenciação dos linfócitos T e B, assim como a função normal das células naturais assassinas. Infecções recorrentes, especialmente fúngicas, são comuns em pacientes com deficiência de biotina.
Rolação da biotina em condições neurológicas de alta dose de biotina-responsiva
Doença basal dos gânglios basais biotínicos (BTBGD) é uma condição neurológica rara que pode se apresentar com convulsões e encefalopatia progredindo para coma e morte. A biotina de alta dose ( 5-10 mg/kg/dia) tem sido usada para tratar com sucesso esta condição, mas o mecanismo de acção é desconhecido.
Recentemente, o tratamento com altas doses de biotina (100-300 mg/dia) tem melhorado os sintomas em um subconjunto de pacientes com esclerose múltipla. Pensa-se que a melhoria dos sintomas neurológicos pode envolver uma melhor produção de mielina secundária ao efeito de altas doses de biotina na síntese de ácidos gordos de cadeia longa.
Role de biotina em indivíduos com distúrbios do cabelo, pele e unhas
Os suplementos de biotina são amplamente utilizados por aqueles que esperam alcançar um cabelo, pele e unhas mais saudáveis. No entanto, há poucas evidências da eficácia da biotina para este uso. Biotina em altas doses tem sido considerada útil em duas condições raras: síndrome do cabelo incombustível familiar e síndrome das unhas quebradiças. Em um estudo com 541 fêmeas que apresentaram queda de cabelo, os níveis séricos de biotina foram encontrados baixos em 38% dos pacientes. O autor concluiu que a etiologia da queda de cabelo é multifactorial e que os suplementos de biotina podem ser considerados se a deficiência de biotina tiver sido demonstrada e outras causas tiverem sido descartadas.
Fontes de Biotina
Biotina está presente em uma grande variedade de alimentos (carnes, laticínios, vegetais, sementes e nozes) e também é produzida por bactérias intestinais. Além disso, uma proporção substancial de indivíduos pode consumir suplementos dietéticos contendo biotina.
Biotina Fisiologia
Ingestada biotina está presente nas formas livre e ligada a proteínas. As formas ligadas às proteínas são digeridas por proteases gastrointestinais e peptidases para formar biocitina e biotina-oligopeptídeos. A biotina livre é liberada da biotimina e biotino-oligopeptídeos pela ação da biotinidase intestinal. A biotina livre é então absorvida no intestino delgado através de um mecanismo mediado por Na+, que também transporta dois outros nutrientes, o ácido pantoténico e o lipoato de sódio e, portanto, é conhecido como o transportador de multivitamínicos dependentes de sódio (SMVT). O gene SMVT humano está localizado no cromossoma 2p23. A actividade do SMVT é regulada pelos níveis de biotina; sendo esta regulada para cima na deficiência de biotina e para baixo na sobre-suplementação de biotina. A biotina de síntese bacteriana está presente na forma não-limitada e é absorvida no intestino grosso por um mecanismo similar mediado por um portador. A produção diária combinada de biotina na urina e fezes excede a ingestão dietética de biotina, sugerindo o importante papel desempenhado pela flora intestinal como fonte de biotina.
Após absorvida, a biotina torna-se disponível para vários processos de biotinilação. A biotina liga-se a cada uma das 5 apocarboxilases para formar a holocarboxilase correspondente (ver imagem abaixo) através da ação da enzima holocarboxilase sintetase.
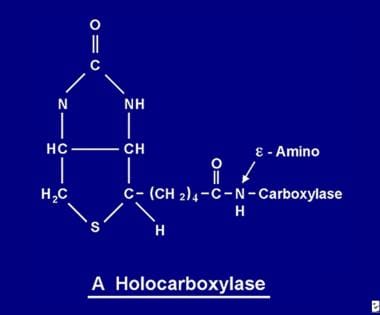 A molécula de biotina é ligada à proteína por uma ligação de peptídeo a um grupo e-amino de uma apocarboxilase para formar uma holocarboxilase.
A molécula de biotina é ligada à proteína por uma ligação de peptídeo a um grupo e-amino de uma apocarboxilase para formar uma holocarboxilase. Reciclagem da biotina
Após a enzima holocarboxilase ter realizado várias carboxilases, ela é capturada por lisossomos celulares. Nos lisossomos, várias enzimas proteolíticas degradam a holocarboxilase para formar a biocitina que, por sua vez, é hidrolisada pela enzima biotinidase para formar a biotina e a lisina. A biotina livre está então disponível para inserção em uma apocarboxilase para formar uma nova molécula de holocarboxilase. Este processo de reciclagem não é 100% eficiente. Como resultado, pequenas quantidades de biotina livre (e alguma biocitina) escapam do ciclo e são perdidas nas fezes e na urina. Por esta razão, a biotina deve ser fornecida ao intestino, para reabastecer a biotina perdida do organismo. As etapas envolvidas na reciclagem da biotina – sua entrada no intestino, sua absorção, sua incorporação nas holocarboxilases, que por sua vez são decompostas para liberar a biotina livre constituem o ciclo da biotina e estão representadas na imagem abaixo.
A enzima biotinidase é essencial para a reciclagem da biotina e portanto os indivíduos com deficiência de biotinidase apresentarão sinais e sintomas de deficiência de biotina.
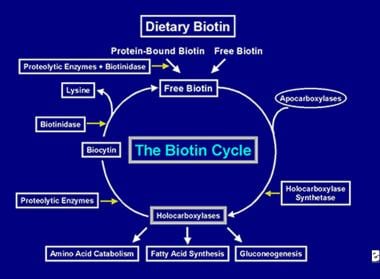 Representação do fluxo de biotina no ciclo da biotina.
Representação do fluxo de biotina no ciclo da biotina. CAUSAS DE DEFICIÊNCIA DE BIOTINA
Como mencionado anteriormente, a biotina está amplamente disponível nos alimentos, é também produzida pela flora intestinal e é amplamente reciclada no organismo com a ajuda da enzima biotinidase; portanto, a deficiência de biotina em indivíduos saudáveis que se alimentam de uma dieta normal é rara. As condições que podem levar à deficiência de biotina são descritas abaixo:
Consumo excessivo de claras de ovo cru: A avidina em claras de ovo cru tem uma alta afinidade com a biotina, tornando-a indisponível para absorção. O calor destrói a avidina, de modo que aqueles que comem ovos cozidos não correm o risco de deficiência de biotina. As claras de ovo cru levam à deficiência de biotina apenas quando consumidas em quantidades excessivas (talvez uma dúzia ou mais por dia).
Nutrição parenteral total sem suplementação de biotina: Vários casos de deficiência de biotina em pacientes que recebem terapia de nutrição parenteral total prolongada (TPN) sem adição de biotina têm sido relatados. Por isso, todos os pacientes que recebem a nutrição parenteral total prolongada também devem receber biotina na dose diária recomendada, especialmente se se espera que a terapia de nutrição parenteral total prolongada dure mais de 1 semana. Actualmente, todas as farmácias hospitalares incluem biotina nas preparações de TPN.
Utilização de fórmulas infantis com biotina inadequada: A deficiência de biotina foi relatada em bebês que receberam fórmulas hipoalergênicas.
Terapia anticonvulsivante crônica: O uso prolongado dos anticonvulsivos, fenobarbital, fenitoína, primidona, e carbamazepina, tem sido ligado à deficiência de biotina. Os mecanismos possíveis incluem a inibição da absorção de biotina pela mucosa intestinal, catabolismo acelerado da biotina e reabsorção renal prejudicada da biotina. Portanto, a biotina suplementar tem sido sugerida para pacientes que são tratados com anticonvulsivos que têm sido ligados à deficiência de biotina.
Terapia antibiótica oral prolongada: O uso prolongado de antibióticos orais tem sido associado à deficiência de biotina. Presume-se que a inibição da flora intestinal que produz biotina seja a base para a carência de biotina. Outro mecanismo possível pode ser o crescimento excessivo de bactérias que consomem biotina, induzido por antibióticos.
Fumar e alcoolismo crônico: Estudos mostraram que fumar pode acelerar o catabolismo da biotina, especialmente nas mulheres. O alcoolismo crônico pode causar a má absorção intestinal da biotina.
Síndrome do intestino curto e doença inflamatória intestinal: Indivíduos com síndrome do intestino curto e doença inflamatória intestinal também estão em risco de deficiência de biotina como resultado da má absorção intestinal de biotina.
Deficiência de biotina marginal durante a gravidez e lactação: Estudos recentes mostraram diminuição dos níveis de biotina em uma proporção significativa de mulheres grávidas e lactantes. Há preocupações de que a deficiência marginal de biotina durante a gravidez possa ser teratogênica e alguns especialistas recomendaram uma maior ingestão de biotina por mulheres grávidas.
Certos erros inatos do metabolismo da biotina também podem levar à manifestação da deficiência de biotina.
A deficiência de biotinidase (BTD) é herdada de forma autossômica recessiva e ocorre com uma freqüência de aproximadamente 1 em 60.000 nascidos vivos; estima-se que 1 em 120 indivíduos seja heterozigoto para a condição. Em indivíduos homozigotos para a doença, os níveis de biotinidase são < 30% normal levando à deficiência de biotina devido à liberação insuficiente de biotina livre devido à diminuição da reciclagem da biotina. Os sintomas de BTD geralmente se desenvolvem entre 1 semana e 1 ano de idade. A gravidade da deficiência da enzima pode variar. Aqueles com deficiência profunda de biotinidase têm níveis de BTD inferiores a 10% do normal, enquanto aqueles com deficiência parcial de biotinidase têm níveis enzimáticos entre 10-30% do normal. Nos EUA e em muitos outros países, a triagem de recém-nascidos inclui testes para deficiência de BTD. Cerca de 150 mutações no gene BTD foram relatadas como causadoras de deficiência de biotinidase. O gene do BTD está localizado no cromossoma 3p25. Um modelo de deficiência de biotinidase em rato foi desenvolvido para estudar vários aspectos da doença.
A deficiência de Holocarboxilase Sintetase (HCLS) também é um distúrbio autossômico recessivo e pode ser diagnosticado prenatalmente. Como discutido anteriormente, a enzima HCLS é necessária para a biotinilação das enzimas apocarboxilase nas formas ativas da holocarboxilase; portanto, a deficiência leva à deficiência de carboxilase múltipla. Os lactentes com esta desordem apresentam nos primeiros meses de vida acidose, hiperamonemia, hipotonia, convulsões e atraso no desenvolvimento. As mutações no gene HCLS causam a deficiência de HCLS.
Porque a deficiência de biotinidase e HCLS leva à diminuição dos níveis das carboxilases dependentes de biotina, as duas condições também foram classificadas como deficiência de carboxilase múltipla. A deficiência profunda de biotinidase era conhecida anteriormente como deficiência de carboxilase múltipla precoce, deficiência parcial de biotinidase como deficiência de carboxilase múltipla tardia ou juvenil e deficiência de HCLS como deficiência neonatal ou de carboxilase múltipla precoce.
Raramente, deficiências isoladas de cada uma das cinco carboxilases individuais dependentes de biotina também podem ocorrer.
A deficiência de biotina devido a um defeito no transporte de biotina também tem sido descrita.
Independentemente da etiologia da deficiência de biotina, as manifestações clínicas são semelhantes. Contudo, a idade de início, as taxas de desenvolvimento dos sintomas e a sequência em que os sintomas aparecem podem ser muito diferentes. Todos os mecanismos responsáveis pelo desenvolvimento das manifestações ainda não foram estabelecidos.