Parole chiave
Ipermobilità, displasia del tessuto connettivo, sindrome di Ehlers-Danlos, scala Beighton, disautonomia, sindrome da tachicardia ortostatica posturale, sindrome dell’intestino irritabile, disturbo di attivazione dei mastociti, displasia articolo-autonomica
Introduzione
Il 20% delle donne e il 10% degli uomini i cui tessuti sono più flessibili e fragili della media sono molto poco riconosciuti e tragicamente maltrattati. Poche opportunità di prevenzione e trattamento superano quella di riconoscere l’ipermobilità, facilmente verificabile chiedendo al paziente di spingere il pollice contro l’avambraccio (Figura 1). Questo segno rivelatore può indicare la strada per una valutazione più dettagliata che è ben all’interno del regno del medico generico, la loro vasta conoscenza clinica che ben si adatta ai problemi multisistemici che possono derivare dal tessuto connettivo lasso. Qui esamino la natura dei disturbi di ipermobilità, dimostro che la conoscenza generale piuttosto che quella sottospecialistica può portare a terapie che cambiano la vita di questi pazienti, e mostro come i test genetici portano descrizioni sintomatiche come la fibromialgia o la sindrome da fatica cronica alla luce perspicace della medicina di precisione.
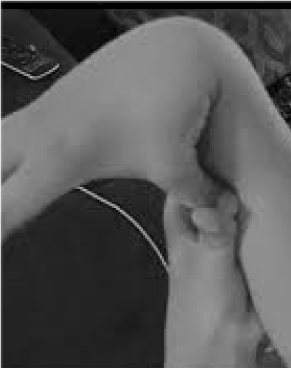
Figura 1. Ipermobilità.
Diverse barriere devono essere superate prima che i pazienti con ipermobilità possano raccogliere i benefici della medicina generale. La prima deriva dall’attenzione pionieristica ma miope dei dermatologi Ehlers e Danlos che hanno impresso l’incredibile ed esotico stiramento della pelle come simbolo fuorviante dei disturbi di lassità del tessuto connettivo. Una seconda barriera è la crescente divisione tra medicina pratica e genetica, la prima opportunamente concentrata sulla malattia comune mentre la seconda fino a poco tempo fa utilizzava la tecnologia specializzata per descrivere il raro. Gli eponimi oscuri con sottotipi e termini tecnici scoraggianti sono come piccole province di un paese straniero poco invitante che nessun viaggiatore globale prenderà tempo per mappare. Contro questi ostacoli c’è un approccio pratico ai disturbi di ipermobilità che si concentra sul riconoscimento tramite anamnesi e visita medica, lasciando i tecnicismi di eponimi, sottotipi e test genetici ai sottospecialisti contattati dopo il primo passo vitale della diagnosi presuntiva. Una volta riconosciuto e riferito, il paziente flessibile rimane dipendente dal suo medico di base, perché solo lui può integrare le raccomandazioni e i risultati dei test nella casa medica generale.
Terminologia
L’aumento della gamma di movimento in più siti di articolazione (articolazione) si verifica in un gran numero di malattie, che vanno dai disturbi cromosomici come la sindrome di Down a disturbi come la sindrome di Ehlers-Danlos (EDS) nota per la loro ipermobilità. McKusick ha reso popolare questa categoria di disturbi noti per la lassità dei tessuti raggruppandoli come disturbi ereditabili del tessuto connettivo, tra cui la sindrome di Marfan su cui si è concentrato come cardiologo. Nel 1977, il dottor Peter Beighton, noto per la sua scala di misurazione della flessibilità, ha organizzato la letteratura intermedia postulando 6-7 tipi di EDS, ora semplificati dalle recenti linee guida come EDS classica, vascolare e ipermobile. Ora è chiaro che questi tipi hanno una considerevole sovrapposizione di sintomi tra di loro e disturbi correlati come l’osteogenesi imperfetta (con numerose fratture), la sindrome di Marfan (dilatazione/dissesto dei grandi vasi), la sindrome di Stickler (cambiamenti oculari e artritici), e molti altri.
Piuttosto che preoccuparsi di malattie particolari, trovo più utile concentrarsi su due principali processi patologici da displasie del tessuto connettivo, illustrate dai molti sintomi elencati nella Tabella 1. Il primo è il dolore articolare da usura (osteoartrite) che differisce dalle articolazioni gonfie e rosse dell’artrite infiammatoria (autoimmune o infiammatoria) che si incontra nell’artrite reumatoide, nel lupus, nella sindrome di Sjogren o nella spondilite anchilosante. Il secondo è lo squilibrio del sistema nervoso autonomo (disautonomia) causato dai vasi sanguigni elastici nei disturbi di ipermobilità . I vasi distensibili causano un ristagno di sangue nel bacino e nelle gambe, con abbassamento della pressione sanguigna e vertigini/stanchezza in piedi (ipotensione ortostatica). Nel tempo, la diminuzione del flusso di sangue alla testa attiva il braccio simpatico del sistema nervoso autonomo (il braccio dell’adrenalina “volo o lotta”), portando a diversi problemi: 1) sindrome da tachicardia ortostatica posturale (POTS) con tachicardia, ansia, stanchezza cronica e “brain fog” (intervalli di diminuzione della concentrazione e della memoria), 2) sindrome del colon irritabile dovuta all’inibizione della motilità intestinale (soppressione parasimpatica) con costipazione alternata a diarrea, gonfiore, reflusso, dolore allo stomaco, disfagia, nausea, e 3) disturbo di attivazione dei mastociti con eruzioni cutanee transitorie, orticaria/pelle reattiva, asma/respirazione affannosa e intolleranze a cibo e farmaci. Ho ideato un termine più ampio che facilita il riconoscimento di qualsiasi disturbo di ipermobilità, riconoscendo le due principali conseguenze della lassità articolare/tessutale: displasia articolo-autonomica o complesso articolare-fatica.
Tabella 1. Risultati tipici e loro frequenza nelle sindromi di ipermobilità. *Da 320 pazienti di sesso femminile e 54 di sesso maschile di età superiore ai 12 anni valutati con questionari dettagliati nel 2017; TMJ, articolazione temporomandibolare; POTS, sindrome da tachicardia ortostatica posturale. Elenco dei sintomi adattato in parte da Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2a ed. Cambridge, MA. Cambridge University Press, 2006.
STORIA (percentuali)*
Infanzia: Coliche (28), difficoltà di alimentazione (19,4); goffo, molte cadute (29,1)
Bambino/adolescente: Aumento di peso lento (12); consapevole della flessibilità (55.6); mostrava trucchi alle articolazioni (53.6)
precoce dolore alle articolazioni (42.1); attività adolescenziali limitate dal dolore-infortunio (55.4); occhiali precoci (46.8)
ortodonzia (61.4%) malattie gengivali (11.3); smalto povero/molte carie (31.6)
Problemi alle articolazioni: Popping (81.6); sublussazioni (62); TMJ popping, catching, dolore (51.2)
dolore in almeno 2 articolazioni (80.3); interventi chirurgici alle articolazioni (33); molte distorsioni (26); fratture >2 (31.5)
ernia del disco spinale (29.3)
Problemi scheletrici: Scoliosi (31); pectus (2.2); andatura in/out (9.3); piedi piatti (15.5)
Pelle: Lividi facili (61); pelle elastica (15); vellutata (18); cicatrici insolite (36.3)
striature prima della gravidanza (73); cicatrizzazione lenta (32.4)
Genitourinario (donne): Menorragia (61,2); endometriosi (11,3); cisti ovariche (55)
sindrome dell’ovaio policistico (8,5); problemi alla vescica (46,1); ernia inguinale/femorale (11,5)
Neuromuscolare: Emicrania (51.7); mal di testa giornaliero (62.3); bisogno di medicine per il mal di testa (45)
Deformazione di Chiari (10.6); neurochirurgia (2.9); intorpidimento/stenosi (48.7); scarso equilibrio (48.6)
dolori neuropatici (37.3); dolori muscolari/spasmi (51.7); debolezza muscolare (32.4)
GI/ombrelli: Costipazione-diarrea (67,1); gonfiore-riflusso-dolore allo stomaco (57,3)
problemi alla cistifellea (12,9); deglutizione difficile (23); nausea frequente (44,9)
Cuore: Prolasso della valvola mitrale (8.7); aneurisma (0.9); aritmia diversa dalla tachicardia (6.8)
POTS: Vertigini in piedi (69,8); sincope (34,5); stanchezza cronica (81,8); difficoltà di sonno (43,5)
brain fog (scarsa memoria o concentrazione a volte-70,8); sensibilità al caldo/freddo (77,5); sudorazione anomala (25); tachicardia (72,8); attacchi di ansia-panico (61,3); voglia di sale (51,2)
Immunità/cellule: Eruzioni cutanee transitorie (33,4%); orticaria/pelle reattiva (45,9%); asma/respirazione affannosa (39,3); intolleranze alimentari/medicinali (61,5%)
Lab/Imaging: Densità ossea bassa (2.1); vitamina D bassa (28.7); vitamina B12 bassa (2.7); ipotiroidismo (11.5); ferritina bassa (1.7); ferro basso (6.7); anticorpo anti-nucleare elevato (7.1)
Numero medio di risultati su 80: femmine >12 (35,4 risultati) maschi >12 (21,5 risultati)
Fisico (percentuale)
Corporatura: Alta-altezza >90° percentile (55.2); pesante-BMI >28 (21.3); magra-BMI <19 (14.2)
Habitus marfanoide (44); aracnodattilia (46.4); segno di Walker-Murdoch presente (40.2)
Faccia: Lunga (25.7); faccia inferiore stretta (5.4); sclera grigio-blu (1.6); palato alto (53.5)
Pelle: Morbida (81,4); traslucida (23,8); sollevamento elastico >Piega di 1 pollice intorno alla mascella su metà avambraccio (51,3)
Le pieghe consistono in uno strato esterno (epidermico) (35.4); cicatrici insolite (42,1%)
Punteggio Beighton: 0-3 punti (8,4); 4-6 punti (32,6); 7-8 punti (38,8); 9 di 9 punti (20,2)
Altre manovre: Unire le mani sopra la spalla-dietro la schiena (68)
segno di preghiera verso l’alto dietro la schiena (71.3); portare la mano intorno alla schiena per toccare l’ombelico (19.5)
Scheletrico: Cifosi del collo (41.2); scoliosi (22.3); lordosi (32.8); piedi piatti (39.6)
andatura: camminare in punta di piedi dentro o fuori (28.3)
Neurologico: Debolezza muscolare (11.2); diminuzione della massa muscolare (8.8)
scarso equilibrio con camminata in tandem (33.4)
Numero medio di reperti su 40: femmine >12 (17.9 reperti) maschi >12 (15.7 risultati)
Displasia articolo-autonomica (AAD)
Un termine come “displasia articolo-autonomica” o AAD può unificare l’approccio ai disturbi di lassità/ipermobilità del tessuto connettivo e contrastare tre problemi che complicano il loro riconoscimento: 1) molti medici e genetisti considerano le displasie del tessuto connettivo come malattie rare ed estreme quando in realtà colpiscono circa l’1% della popolazione 2) le linee guida diagnostiche dell’EDS, pur semplificando i principali tipi di EDS come classica, vascolare e ipermobilità, confondono ancora facendo riferimento a molti tipi minori che formano uno spettro di sintomi sovrapposti, e 3) la disautonomia quasi obbligatoria che accompagna l’EDS e i disturbi correlati (Tabella 1) è omessa come criterio diagnostico, analogamente alla descrizione di un elefante senza menzionarne la proboscide. La DAA denota l’inevitabile connettivo fisiologico tra il tessuto lasso e la disautonomia nello stesso modo in cui le sindromi metaboliche o epatorenali descrivono come l’obesità porta alla malattia epatica/diabete o l’insufficienza epatica causa la malattia renale.
Come la maggior parte dei modelli medici o sindromi, compresa la EDS, la DAA è un complesso di sintomi con molte cause sottostanti. “Articulo” si riferisce all’articolazione, riconoscendo che l’afflizione di qualsiasi componente articolare – pelle, nervi, muscoli, ossa, tessuto connettivo, vasi sanguigni – causa un modello simile di lassità articolare con squilibrio autonomo. Ci sarà fragilità della pelle, diminuzione della densità ossea con suscettibilità alle fratture, debolezza muscolare con dolore (mialgia), neuropatia con intorpidimento, tunnel carpale o sindromi dolorose regionali, cambiamenti autonomi tra cui tachicardia, ansia, sonno non ristoratore, immotilità intestinale, orticaria, intolleranze alimentari e medicinali, tutti presenti in misura variabile anche all’interno di specifici “tipi” di EDS. Non ho trovato grandi differenze tra i pazienti che si adattano a particolari tipi di EDS, come illustrato da cambiamenti cutanei più frequenti nella cEDS classica di tipo I contro una maggiore flessibilità articolare nella hEDS ipermobile di tipo III. Le frequenze dei sintomi in 374 pazienti registrati tramite interrogazione sistematica non variavano significativamente tra quelli che ho diagnosticato come cEDS contro hEDS, e anche gli 8 pazienti identificati come possibili vEDS vascolari avendo mutazioni del gene del collagene tipo III non erano distinti (gli aneurismi presenti in uno degli 8 pazienti avevano la stessa bassa frequenza in altre categorie EDS). La cosa più importante è il concetto di uno spettro EDS, meglio rappresentato a mio parere come AAD.
Il riconoscimento del complesso di sintomi AAD, causato dalla EDS e dalle displasie del tessuto connettivo correlate, eviterà ai pazienti tristi viaggi da uno specialista all’altro, diagnosi sintomatiche come vertigini, svenimenti, fibromialgia o stanchezza cronica, e tragiche sofferenze quando molte strategie preventive e trattamenti sono disponibili. Inoltre, il riconoscimento della disautonomia spiega l’ansia da POTS e la depressione che frequentemente accompagna il dolore cronico o la limitazione dell’attività, differenze di comportamento che possono essere riconosciute come conseguenze della malattia medica piuttosto che psichiatrica.
I test genetici supportano il concetto di displasia articolo-autonomica
Implicato dalla loro caratterizzazione come disturbi ereditabili del tessuto connettivo, le forme più estreme di EDS e disturbi correlati sono stati trovati per esibire eredità autosomica dominante, nel senso che gli individui affetti hanno una forma normale e una anormale del gene (allele). L’eredità dominante ha senso perché la proteina difettosa prodotta dall’allele anormale, analoga a un mattone deformato, si mescolerebbe con i mattoni normali dell’altro allele e creerebbe una struttura traballante. Il riconoscimento dell’articolazione come una struttura multicomponente, vincolata dalla pelle sovrastante, sostenuta dall’osso interno e dalla cartilagine, e che agisce attraverso nervi intrinseci, muscoli, tendini, rivestimento articolare e vasi, implica a sua volta che uno qualsiasi di questi elementi può essere colpito per produrre una displasia, un’alterazione del tessuto distinta dalle malformazioni causate da altri disturbi genetici. Lo sviluppo della clonazione e del sequenziamento del DNA confermerebbe il funzionamento dell’eredità autosomica dominante e la capacità dei geni che alterano qualsiasi elemento della struttura articolare di causare il quadro sintomatologico della DAA.
I test genetici iniziarono con un focus su un gene “candidato” selezionato a causa dei sintomi del paziente, illustrato dal test del gene fibrillina-1 per la sindrome di Marfan. Sequenziare le lettere AGCT del DNA del gene, il “testo” dei nucleotidi, potrebbe rivelare sostituzioni o “typos” che alteravano l’aminoacido della proteina codificata e la sua funzione prevista. Poi è arrivata la nuova tecnologia chiamata NextGen o sequenziamento parallelo rapido del DNA, analogo alla lettura di tutte le pagine di un libro in una sola volta piuttosto che da un capo all’altro. Questo ha permesso il sequenziamento di gruppi di geni (pannelli) come quelli che causano la cardiomiopatia o la displasia del tessuto connettivo o anche tutti i 23.000 geni in ogni cellula umana (il nostro genoma) in 2-3 mesi. Il sequenziamento delle regioni genetiche codificanti le proteine o esoniche, chiamato sequenziamento dell’esoma intero (WES), può essere ordinato da molte aziende e nella mia pratica è stato ottenuto per meno di $ 200-300 di costo di tasca propria nella metà dei miei pazienti (prezzo di listino è $ 9000 e l’importo fatturato all’assicurazione è di circa $ 20.000).
Anche se interpretare la media di 12.000 varianti del DNA dettagliate da WES rimane un problema, diminuito da una crescente epidemiologia di varianti nelle popolazioni normali e malattia, rendimenti di cambiamenti genici che contribuiscono ai sintomi AAD tra 620 pazienti sottoposti a WES nella mia pratica è oltre il 60%. Oltre alle oltre 80 mutazioni dei geni del collagene, comprese quelle del collagene di tipo I solitamente associato all’osteogenesi imperfetta, del collagene di tipo V spesso associato alla EDS classica, dei collageni di tipo VI, IX e XII inizialmente descritti con le miopatie e del temuto collagene di tipo III associato all’EDS vascolare, sono ricorrenti mutazioni nei geni che interessano i componenti cutanei, nervosi, muscolari, fibrillari e vascolari che rafforzano un concetto di DAA in cui l’alterazione di qualsiasi componente articolare produce il modello di segni e sintomi descritto nella tabella 1. Ora il riconoscimento dell’EDS e dei disturbi correlati inclusi nel concetto di DAA può rafforzare l’impressione clinica con la prova oggettiva del DNA che permette la consulenza del paziente e della famiglia.
Diagnosi e terapia dei disturbi di ipermobilità/AAD
L’attenzione per i sintomi principali di dolore/fragilità articolare e autonomica (intestino, cuore, disturbi immunitari) può portare a test rapidi per l’ipermobilità come mostrato nella Figura 1. Il segno del pollice all’avambraccio può prefigurare l’intera scala di Beighton, dando un punto bilateralmente per le dita premute indietro oltre i 90 gradi sulla mano, toccando i pollici agli avambracci, iperestendendo il gomito con le braccia estese, iperestendendo le ginocchia per formare una curva della gamba all’indietro, e 1 punto per la capacità di toccare i palmi al pavimento . Una breve indagine dei sintomi come quelli della tabella 1 può confermare il sospetto, aiutata forse includendo negli esami fisici alcune delle valutazioni elencate nella parte inferiore della tabella 1. Si noti che non tutti i pazienti affetti da DAA sono alti o magri, e che una parte sostanziale sarà in sovrappeso a causa delle restrizioni di attività dovute al dolore e alla fatica. Molti avranno la pelle elastica, esaminata tirando su le pieghe intorno alla mascella o sull’avambraccio, in particolare quando queste sono le pieghe sottili e superficiali dell’epidermide. L’accumulo di tessuto bianco o la formazione di cheloidi intorno alle cicatrici è comune, e la valutazione di Beighton o altre manovre di ipermobilità elencate nella tabella 1 possono essere utili. Si può notare una postura afflosciata con curva del collo in avanti, scoliosi e lordosi, insieme a piedi piatti o alluci in fuori (più che in dentro) quando si cammina. La forza muscolare sarà in calo in alcuni pazienti insieme a uno scarso equilibrio nella camminata in tandem, ma il generalista con un tempo limitato ha bisogno di occuparsi solo di alcuni di questi segni fisici; il rinvio stabilirà il resto.
Il più importante sarà l’ortopedico che ha familiarità con l’osteoartrite e i cambiamenti associati all’EDS, come i cambiamenti degenerativi delle articolazioni, la cartilagine che si deteriora, le bande di plica e le regioni di osso morto. Un ortopedico esperto e conservatore che riconosce gli scarsi risultati degli interventi chirurgici per “stringere” o sostituire le articolazioni è auspicabile. Un cardiologo può documentare la POTS usando il tilt-table test per i cambiamenti ortostatici nella frequenza cardiaca e nella pressione sanguigna, eseguendo anche studi elettro ed ecocardiografici per escludere la dilatazione aortica o gli aneurismi. La neurologia può valutare le comuni emicranie o cefalee quotidiane, la deformazione di Chiari che si verifica con lo slittamento e l’erniazione del tessuto cerebrale lasso, la neuropatia dovuta alla costrizione all’interno delle articolazioni lasse, e i cambiamenti muscolari denotati dalla descrizione del sintomo fibromialgia ed evidenziati dai frequenti cambiamenti genetici miopatici che sta promuovendo un nuovo termine di EDS miopatica. Gli allergologi che hanno familiarità con l’attivazione dei mastociti e i gastroenterologi che possono documentare la bassa motilità intestinale sono aggiunte preziose al team della sottospecialità, e nessuno è più prezioso del medico di base per gestire le molte raccomandazioni e i farmaci che possono essere richiesti. Anche se può sembrare strano detto da un genetista, penso che le valutazioni di sottospecialità che portano alla terapia siano più importanti della valutazione e dei test genetici, ma questi ultimi possono essere utili per disinnescare le supposizioni sulla malattia psichiatrica.
Il trattamento inizia con un programma di esercizi che può giovare sia all’artrite dovuta all’ipermobilità articolare che alla sua disautonomia riflessiva. L’attenzione ai dolori articolari (i dolori di crescita si manifestano di notte) o alle lesioni/deformità scheletriche può istituire una prevenzione rendendo il paziente consapevole della sua condizione (notare nella tabella 1 che molti non riconoscono la loro flessibilità come anormale). Le lesioni da usura possono essere prevenute rinforzando le articolazioni suscettibili, con fasce in vita per sostenere la schiena, e scegliendo attività che proteggono le articolazioni come il nuoto piuttosto che il jogging. I pazienti dovrebbero rimanere attivi con un moderato sollevamento pesi e altre attività ragionevoli per costruire i muscoli intorno alle articolazioni, evitando cicli di inattività che esacerbano la fatica cronica o la fibromialgia. La terapia fisica appropriata, la gestione del dolore, la guida all’esercizio e i farmaci aneddotici come i preparati di glucosamina/condroitina hanno aiutato molti, ma i rilassanti che tolgono la protezione muscolare delle articolazioni e le iniezioni di steroidi che mascherano le lesioni, promuovono l’osteoporosi/degenerazione e danno solo un sollievo temporaneo dovrebbero essere evitati.
Il pilastro della terapia della disautonomia è l’idratazione (8-10 bicchieri di liquido al giorno), il sale (ipertensione labile assente), e una dieta ad alto contenuto proteico, tutti volti ad aumentare il volume dei fluidi nel sangue e migliorare la circolazione cerebrale. Una terapia nutrizionale supplementare può includere vitamina C (2 g al giorno) vitamina D (>2000 unità al giorno, più delle 400-800 dei multivitaminici), vitamina B12 (1-2,5 mg al giorno). La bassa motilità intestinale o gastroparesi viene aiutata evitando i liquidi prima dei pasti, pasti più piccoli e frequenti, probiotici, preparati enzimatici e, a detta di molti, meno glutine e latticini che sono irritanti comuni. Programmi di esercizio in posizione supina, biofeedback, e molte opzioni di farmaci sono disponibili attraverso i cardiologi per POTS (ad esempio, beta-bloccanti, Midodrine, Florinef), attraverso neurologi o esperti di gestione del dolore (ad esempio, Gabapentin, Lyrica), o attraverso allergologi che possono fornire protocolli antistaminici per sopprimere i mastociti (ad esempio, Zyrtec, Zantac, Cromolyn, Singulair). I vantaggi dei test genomici come il WES che va oltre i geni noti per causare la displasia del tessuto connettivo sono nuovi risultati come i geni legati ai muscoli ora noti per causare EDS e la documentazione di risultati accidentali o secondari come le mutazioni del gene del cancro al seno e alle ovaie. La scoperta di geni associati a sintomi più gravi permette ai pazienti di conoscere i rischi per la prole e di scegliere strategie di diagnosi genetica prenatale o preimpianto compatibili con i loro valori.
Presentazione del caso
Eve è venuta dal suo medico all’età di 18 anni, vertigini, svenimenti, ansia e mal di stomaco, così esausta che si è ritirata il venerdì sera per riposare tutto il weekend. Era stata sana, attiva nella ginnastica e nel tifo, una ragazza coraggiosa e brillante, la cui perseveranza nonostante gli infortuni le fece guadagnare gli applausi degli allenatori. Poi l’influenza dalla quale non si è più ripresa, la nausea così forte che non poteva mangiare, la perdita di peso, lo svenimento in classe, il risveglio notturno con il cuore che batteva, il risveglio al mattino senza sonno ristoratore. Eve ha visto molti medici, anche al pronto soccorso quando le palpitazioni e il panico le toglievano il respiro. Non c’era nulla di sbagliato, tranne lo stress, dicevano, tutto nella sua mente. Calmata ma non curata dalle pillole, con il lavoro che veniva meno per la fatica e la confusione, si chiedeva se stesse impazzendo. Non veniva riconosciuta la sua pelle morbida e fragile, le sue mestruazioni dure e pesanti, i suoi mal di testa quotidiani e, cosa più sorprendente di tutte, l’ipermobilità che lei e la sua famiglia supponevano fosse normale: “Sì, siamo piegati e con due articolazioni, non lo siamo tutti? Eve avrebbe risposto se le fosse stato chiesto.
Fortunatamente Eve cercò un nuovo medico di base che fu in grado di documentare molti dei segni e dei sintomi elencati nella Tabella I. Eve aveva coliche precoci e reflusso con difficoltà ad allattare, era molto flessibile, chiamata dalla madre la bambina “senza ossa”, e quando era più grande si sedeva sulle ginocchia con le gambe distese dietro di lei (la posizione “W”), dicendo che era più comoda. All’inizio camminava lentamente, cadendo spesso e apparendo goffa, ma presto fu attiva e si arrampicava ovunque, facendo trasalire i suoi genitori mettendole un piede al collo o facendo la spaccata con facilità. Ma quando entrò nella scuola elementare cominciò a lamentarsi dell’artralgia, le gambe le facevano male di notte e le ginocchia e le caviglie le davano fastidio dopo l’attività. Il suo pediatra rassicurò i suoi genitori che stava solo avendo “dolori di crescita”, ma si preoccuparono di più quando cercò di giocare a calcio e continuò a girare le caviglie, una distorsione richiese uno stivale per 2 mesi dopo che fu notata una frattura da stress.
Eve dovette smettere di giocare a calcio ma scoprì di eccellere nella ginnastica man mano che cresceva, scegliendo esercizi a terra a causa dello scarso equilibrio sulla trave e una tendenza della sua spalla a tirare sulle barre parallele. Notò che le sue articolazioni scoppiavano con il movimento e spesso le faceva scoppiare di proposito perché sembrava alleviare la tensione e il dolore. Provò a fare la cheerleader con successo nelle manovre, ma scoprì di essere troppo sensibile al calore. Le altre ragazze la chiamavano “faccia di pomodoro” perché si surriscaldava durante le routine, e si stancava facilmente, avendo spesso bisogno di un giorno di riposo dopo l’esercizio. I suoi problemi digestivi riemersero con frequenti costipazioni e dolori allo stomaco, spesso si sentiva gonfia e aveva bisogno di lassativi. In sesta classe saltava la scuola a causa dei suoi problemi intestinali e dei dolori articolari, sentendosi spesso affaticata e stordita. Non si sentiva riposata al mattino e spesso faceva dei sonnellini nei fine settimana, un allontanamento dal suo precedente stile di vita attivo ed entusiasta. Si sentiva spesso ansiosa, senza essere in grado di risalire ad alcuno specifico stress scolastico o familiare, e cominciò ad avere mal di testa al mattino che pensava fossero dovuti al suo scarso sonno. I suoi genitori la portarono di nuovo dal pediatra e le dissero di nuovo che non c’era niente che non andava, ma il solito stress scolastico.
Quando aveva 14 anni Eve ebbe una brutta malattia simile all’influenza e perse la scuola per un mese con grave affaticamento, mal di testa, nausea, vomito e perdita di peso. Aveva terribili vertigini e alcuni dolori al petto, sentendo che il suo cuore batteva a volte (avendo palpitazioni). I suoi genitori chiesero consiglio a un altro pediatra, che mandò Eve da un cardiologo, il quale pensò che avesse una tachicardia dovuta allo stress e all’ansia per la sua malattia. Egli diede a Eve il propranololo, un beta-bloccante, per rallentare la sua frequenza cardiaca, ma lei divenne ancora più affaticata e ora era costretta a letto per gran parte della giornata. Il suo dolore alle articolazioni aumentò dopo la malattia e lei si attardava a letto per la maggior parte del giorno, sentendosi triste e diventando ansiosa quando i suoi genitori cercavano di portarla fuori o di farle fare un giro in macchina. La riportarono dal secondo pediatra che la indirizzò alla psichiatria, e lo Zoloft fu iniziato per aiutarla con l’ansia e la depressione. Dopo pochi giorni Eve scoppiò in un’eruzione cutanea con orticaria e dovette smettere quel farmaco. Continuò ad avere vertigini e fu visitata da altri due specialisti, un gastroenterologo che pensava che potesse avere la celiachia e un otorinolaringoiatra che diagnosticò le vertigini e la mandò a neurologia. Raccontò a quel medico dei mal di testa provenienti dalla parte posteriore della testa, e fu ottenuto uno studio di risonanza magnetica in posizione supina che era normale.
L’apprezzamento della probabile diagnosi di DAA da parte del suo medico di base istituì un diverso giro di rinvii di sottospecialità, compresa la neurologia dove uno studio di risonanza magnetica in posizione verticale dimostrò l’erniazione della deformazione di Chiari. Un cardiologo ha documentato un test tilt-table anormale insieme a una bassa motilità intestinale e ha dimostrato che la sua ansia e l’affaticamento da stress erano dovuti alla disautonomia. Le strategie di idratazione nutrizionale, sale e proteine con una terapia a base di beta-bloccanti e antistaminici migliorarono la sua fatica e i suoi sintomi intestinali e diminuirono i suoi mal di testa. Fu deciso che l’ernia di Chiari di 4-5 mm non era sufficiente per richiedere un intervento chirurgico, e la sua gestione psichiatrica fu riconosciuta come il trattamento dei sintomi secondari di un disturbo medico. Alcuni dei suoi sottospecialisti, scettici sulla sua diagnosi clinica, furono convinti dai test genetici che documentarono un cambiamento in un gene per la catena alfa-1 del collagene di tipo V, abbreviato COL5A1, un risultato comune nei pazienti con EDS. Eve aveva ereditato la mutazione da sua madre, che aveva avuto sintomi più lievi di EDS e ora era in grado di gestirli meglio.
Nel corso di diversi mesi la salute e la vitalità di Eve furono ripristinate e lei “riavrà la sua vita”, come direbbero lei e i suoi genitori. Aveva ancora dolori articolari, ansia e stanchezza a volte, ma il “saggio esercizio” per costruire una protezione muscolare intorno alle sue articolazioni, gli approcci nutrizionali per la disautonomia e gli approcci di biofeedback per prevenire l’ansia le cambiarono la vita. Questi trattamenti sono stati tutti dovuti al riconoscimento da parte del suo medico di famiglia, ora in grado di coordinare le cure per Eve e sua madre e di fornire risorse per la comprensione sia della EDS che dei suoi sottostanti cambiamenti genetici. Il passaggio alla generazione successiva è stata la capacità di informare Eve del suo rischio del 50% di trasmettere la sua mutazione COL5A e alcuni rischi di travaglio precoce e sanguinamento postpartum in qualsiasi gravidanza futura.
- Remvig L, Jensen DV, Ward RC (2007) Epidemiologia di ipermobilità articolare generale e la base per i criteri proposti per la sindrome di ipermobilità articolare benigna: revisione della letteratura. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) La sindrome di Ehlers-Danlos. In: Il tessuto connettivo e i suoi disordini ereditabili. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Parte C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Disautonomia nella sindrome di ipermobilità articolare. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lezioni di terapia del dolore – una personale esperienza post-laurea. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) Una piccola molecola per una grande malattia. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Capitolo 11: sequenziamento del gene e del genoma: Interpretare la variazione genetica a livello di nucleotide. In: Variazione cromosomica umana: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Sequenziamento dell’esoma diagnostico – ci siamo già? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Riscrivere la storia naturale del dolore e dei sintomi correlati nella sindrome di ipermobilità articolare/Ehlers-Danlos, tipo ipermobilità. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Sindrome di ipermobilità articolare benigna: valutazione, diagnosi e gestione. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) La sindrome di ipermobilità. Disturbi muscoloscheletrici associati all’ipermobilità articolare generalizzata. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) diagnosi genetica presintomatica e preimpianto: Neurologia, NextGenetics e la prossima generazione. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Gestione del dolore e della fatica nella sindrome da ipermobilità articolare (nota anche come sindrome di Ehlers-Danlos, tipo ipermobilità): principi e proposta di un approccio multidisciplinare. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Riscrivere la storia naturale del dolore e dei sintomi correlati nella sindrome di ipermobilità articolare/Ehlers-Danlos, tipo ipermobilità. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Trattamento multidisciplinare della disabilità nella sindrome di Ehlers-Danlos tipo ipermobilità/sindrome di ipermobilità: uno studio pilota utilizzando una combinazione di terapia fisica e cognitivo comportamentale su 12 donne. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exome analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am J Med Genet A 164A: 1209-1212.