Palavras-chave
hipermobilidade, displasia do tecido conjuntivo, síndrome de ehlers-danlos, escala de beighton, disautonomia, síndrome de taquicardia ortostática postural, síndrome do intestino irritável, distúrbio de ativação dos mastócitos, displasia articulo-autonômica
Introdução
Muito pouco reconhecido e tragicamente maltratado são os 20% das mulheres e 10% dos homens cujos tecidos são mais flexíveis e frágeis do que a média . Poucas oportunidades de prevenção e tratamento excedem a de reconhecer a hipermobilidade, facilmente rastreada ao pedir ao paciente que empurre o polegar contra o antebraço (Figura 1). Este sinal de conto pode indicar o caminho para uma avaliação mais detalhada que está bem dentro do domínio do médico de clínica geral, o seu amplo conhecimento clínico combinando bem com os problemas multissistémicos que podem surgir do tecido conjuntivo laxante. Aqui revejo a natureza dos distúrbios de hipermobilidade, demonstro que o conhecimento geral e não subespecializado pode levar a terapias que mudam a vida destes pacientes, e mostro como os testes genéticos trazem descrições sintomáticas como fibromialgia ou síndrome de fadiga crônica para a luz perspicaz da medicina de precisão.
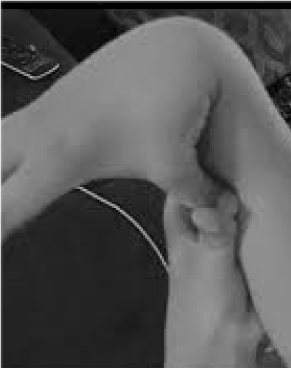
Figura 1. Hipermobilidade.
Barreiras transversais devem ser superadas antes que os pacientes com hipermobilidade possam obter os benefícios da medicina geral. A primeira deriva envolve o foco pioneiro mas míope dos dermatologistas Ehlers e Danlos imprinting incrível e exótico estiramento da pele como o símbolo enganador de distúrbios de frouxidão do tecido conjuntivo. Uma segunda barreira é a crescente divisão entre medicina prática e genética, a primeira concentrando-se apropriadamente na doença comum, enquanto a segunda até há pouco tempo usava tecnologia especializada para descrever as raras. Os obscuros epónimos com subtipos assustadores e termos técnicos são como pequenas províncias de um país estrangeiro pouco convidativo que nenhum viajante global levará tempo a mapear. Contrariar estes obstáculos é uma abordagem prática às doenças de hipermobilidade que se concentra no reconhecimento pela história e física, deixando os aspectos técnicos dos eponyms, subtipos e testes genéticos para subspecialistas contactados após o primeiro passo vital do diagnóstico presuntivo. Uma vez reconhecido e encaminhado, o paciente flexível permanece dependente do seu médico primário, pois apenas ele pode integrar recomendações e resultados de testes no lar médico geral.
Terminologia
Ocorre uma amplitude de movimento aumentada em múltiplos locais de articulação (articulação) num grande número de doenças, desde doenças cromossómicas como a síndrome de Down a doenças como a síndrome de Ehlers-Danlos (SED) conhecida pela sua hipermobilidade. McKusick popularizou esta categoria de doenças conhecidas pela frouxidão dos tecidos, agrupando-as como doenças hereditárias do tecido conjuntivo, incluindo a síndrome de Marfan, na qual se concentrou como cardiologista. Em 1977, o Dr. Peter Beighton, conhecido por sua flexibilidade de medição em escala, organizou a literatura interventiva postulando 6-7 tipos de SED, agora simplificados por diretrizes recentes como SED clássica, vascular e hipermóvel. Agora está claro que esses tipos têm considerável sobreposição de sintomas entre si e distúrbios relacionados como osteogênese imperfeita (com numerosas fraturas), síndrome de Marfan (dilatação/dissecção de grandes vasos), síndrome de Stickler (alterações nos olhos e artrite) e muitos outros.
Cuidado com doenças particulares, acho mais útil focar em dois processos de doenças principais a partir de displasias do tecido conjuntivo, ilustradas pelos muitos sintomas listados na Tabela 1. Primeiro é a dor articular de desgaste (osteoartrite) que difere das articulações inchadas e vermelhas da artrite inflamatória (auto-imune ou inflamatória) encontrada na artrite reumatóide, lúpus, síndrome de Sjogren, ou espondilite anquilosante. Segundo é o desequilíbrio do sistema nervoso autônomo (disautonomia) causado por vasos sanguíneos elásticos em distúrbios de hipermobilidade . Os vasos distensíveis causam a acumulação de sangue na pélvis e nas pernas, com pressão sanguínea mais baixa e tonturas/fainidades em pé (hipotensão ortostática). Com o tempo, a diminuição do fluxo sanguíneo para a cabeça activa o braço simpático do sistema nervoso autonómico (o braço de “fuga ou luta” de adrenalina), levando a vários problemas: 1) síndrome de taquicardia ortostática postural (STPO) com taquicardia, ansiedade, fadiga crônica e “neblina cerebral” (intervalos de redução do foco e da memória), 2) síndrome do intestino irritável devido à inibição da motilidade intestinal (supressão parassimpática) com a constipação alternada com diarréia, inchaço, refluxo, dor de estômago, disfagia, náusea e 3) distúrbio de ativação de mastócitos com erupções cutâneas transitórias, urticária/ pele reativa, asma/exalação intensa e intolerâncias alimentares/medicamentos. Eu concebi um termo mais amplo que facilita o reconhecimento de qualquer distúrbio de hipermobilidade, reconhecendo as duas principais conseqüências da frouxidão articular/teticular: displasia articulo-autonômica ou complexo junção-fadiga.
Tabela 1. Achados típicos e sua freqüência em síndromes de hipermobilidade. *A partir de 320 pacientes do sexo feminino e 54 do masculino acima de 12 anos de idade avaliados por questionários detalhados em 2017; ATM, articulação temporomandibular; POTS, síndrome taquicardia ortostática postural. Lista de sintomas adaptada em parte da Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providenciando um lar médico. 2ª ed. Cambridge, MA. Cambridge University Press, 2006.
HISTÓRIA (percentagens)*
Infância: Cólicas (28), dificuldades de alimentação (19,4); desajeitadas, muitas quedas (29,1)
Criança/teen: Ganho de peso lento (12); atento à flexibilidade (55,6); mostrou truques articulares (53,6)
dores articulares precoces (42,1); atividades adolescentes limitadas pela dor-lesão (55,4); óculos precoces (46,8)
ortodontia (61,4%) doença gengival (11,3); esmalte pobre/muitas cáries (31,6)
Problemas de articulação: Rebentamento (81,6); subluxações (62); ATM rebentando, pegando, dor (51,2)
dor em pelo menos 2 articulações (80,3); cirurgias articulares (33); muitas entorses (26); fraturas >2 (31,5)
hérnia de disco vertebral (29,3)
questões esqueléticas: Escoliose (31); pectus (2.2); in/out (9.3); pés chatos (15.5)
Skin: Contusões fáceis (61); pele elástica (15); aveludada (18); cicatrizes incomuns (36,3)
estrias antes da gravidez (73); cicatrização lenta (32,4)
Geniturinário (fêmeas): Menorragia (61,2); endometriose (11,3); cistos ovarianos (55)
síndrome ovariana policística (8,5); problemas urinários (46,1); hérnia inguinal/femoral (11,5)
Neuromuscular: Enxaquecas (51,7); dores de cabeça diárias (62,3)
Deformação de Chiari (10,6)
Dores de tiro neuropático (37,3)
GI/bowels: Constipação-diarreia (67,1); dores de inchaço e refluxo-estômago (57,3)
problemas de vesícula biliar (12,9); deglutição difícil (23); náuseas frequentes (44,9)
Coração: Prolapso da válvula mitral (8,7); aneurisma (0,9); arritmia que não taquicardia (6,8)
POTS: Tonturas em pé (69,8); síncope (34,5); fadiga crônica (81,8); dificuldades de sono (43,5)
neblina cerebral (memória fraca ou foco às vezes-70,8); sensibilidade ao calor/frio (77,5); sudorese anormal (25); taquicardia (72,8); ataques de ansiedade-pânico (61,3); fantasia de sal (51,2)
Imune/célula mastro: Erupções transitórias (33,4%); urticária/pele reactiva (45,9%); asma/exalação (39,3); intolerâncias à medicação alimentar (61,5%)
Lab/Imaging: Baixa densidade óssea (2,1); baixa vitamina D (28,7); baixa vitamina B12 (2,7); hipotiroidismo (11,5); baixa ferritina (1,7); baixo ferro (6,7); elevado anticorpo antinuclear (7.1)
A média de achados em 80: fêmeas >12 (35,4 achados) machos >12 (21,5 achados)
Físico (porcentagem)
Build: Tall-height >90º percentil (55,2); IMC pesado >28 (21,3); IMC fino <19 (14,2)
Hábito marfanoide (44); aracnodactilia (46,4); sinal de Walker-Murdoch presente (40,2)
Face: Longo (25,7); face inferior apertada (5,4); esclerótica azul-cinza (1,6); palato alto (53,5)
Pele: Macia (81,4); translúcida (23,8); dobra elástica >1 polegada em torno da mandíbula no meio da exploração (51,3)
As dobras consistem em camada externa (epidérmica) (35.4); cicatrizes pouco usuais (42,1%)
Pontuação de peso: 0-3 pontos (8,4); 4-6 pontos (32,6); 7-8 pontos (38,8); 9 de 9 pontos (20,2)
Outras manobras: Juntar as mãos sobre as costas (68)
Sinal de oração para cima atrás das costas (71,3); trazer a mão para trás para tocar umbigo (19,5)
>
Esqueleto: Cifose cervical (41,2); escoliose (22,3); lordose (32,8); pés chatos (39,6)
>
andar para dentro ou para fora (28,3)
Neurológico: Fraqueza muscular (11,2); diminuição da massa muscular (8,8)
mau equilíbrio por caminhada tandem (33,4)
A média de achados em 40: fêmeas >12 (17,9 achados) machos >12 (15.7 achados)
Displasia Articulo-Autonômica (DAA)
Um termo como “displasia articulo-autonômica” ou DAA pode unificar a abordagem aos distúrbios de frouxidão do tecido conjuntivo/hipermobilidade e combater três problemas que complicam o seu reconhecimento: 1) muitos médicos e geneticistas vêem as displasias do tecido conjuntivo como doenças raras e extremas quando na realidade afectam cerca de 1% da população 2) as orientações diagnósticas da SED, embora simplificando os principais tipos de SED como clássica, vascular e hipermobilidade, ainda confundem ao referir-se a muitos tipos menores que formam um espectro de sintomas sobreposto, e 3) a quase obrigatoriedade da disautonomia que acompanha a SED e doenças relacionadas (Tabela 1) é omitida como critério diagnóstico, análogo à descrição de um elefante sem mencionar a sua tromba. AAD denota a inevitável ligação fisiológica entre tecido solto e disautonomia da mesma forma que as síndromes metabólicas ou hepatorrenais descrevem como a obesidade leva a doença hepática/diabetes ou insuficiência hepática causa doença renal.
Como a maioria dos padrões médicos ou síndromes, incluindo a SED, a DAA é um complexo de sintomas com muitas causas subjacentes. “Articulo” refere-se à articulação, reconhecendo que a aflição de qualquer componente articular – pele, nervo, músculo, osso, tecido conjuntivo, vaso sanguíneo – causa um padrão similar de frouxidão do tecido conjuntivo com desequilíbrio autonômico. Haverá fragilidade da pele, diminuição da densidade óssea com susceptibilidade a fracturas, fraqueza muscular com dor (mialgia), neuropatia com entorpecimento, túnel do carpo ou síndromes regionais de dor, alterações autonómicas incluindo taquicardia, ansiedade, sono não-restaurante, immotilidade intestinal, urticária/lesões, intolerâncias de medicação alimentar, tudo isto presente em extensão variável mesmo dentro de “tipos” específicos de SED. Não encontrei grandes diferenças entre os pacientes que se enquadram em determinados tipos de SEDE, ilustradas pelas alterações cutâneas mais frequentes nos SEDE clássicos do tipo I versus uma maior flexibilidade articular nos SEDE hipermóveis do tipo III. As frequências de sintomas em 374 pacientes registrados por consulta sistemática não variaram significativamente entre os pacientes I diagnosticados como SEDE versus SEDE, e mesmo os 8 pacientes identificados como possíveis vEDS vasculares por apresentarem mutações do gene colágeno tipo III não eram distintos (os aneurismas presentes em um dos 8 pacientes tinham a mesma baixa frequência em outras categorias de SEDE). O mais importante é o conceito de um espectro de SED, melhor representado na minha opinião como DAA.
O reconhecimento do complexo de sintomas da DAA, causado pela SED e displasias do tecido conjuntivo relacionadas, evitará viagens tristes de especialista para especialista, diagnósticos sintomáticos como vertigens, desmaios, fibromialgia ou fadiga crônica, e sofrimento trágico quando muitas estratégias e tratamentos preventivos estão disponíveis. Além disso, o reconhecimento da disautonomia explica a ansiedade do POTS e a depressão que frequentemente acompanha a dor crônica ou limitação da atividade, diferenças de comportamento que podem ser reconhecidas como conseqüências de doenças médicas e não psiquiátricas.
Testes genéticos suportam o conceito de displasia articulo-autonômica
Implicado por sua caracterização como distúrbios hereditários do tecido conjuntivo, as formas mais extremas de SED e distúrbios relacionados foram encontradas para exibir herança autossômica dominante, significando que os indivíduos afetados têm uma forma normal e uma forma anormal do gene (alelo). A herança dominante faz sentido porque a proteína defeituosa produzida pelo alelo anormal, análoga a um tijolo deformado, se misturaria com tijolos normais do outro alelo e faria uma estrutura instável. O reconhecimento da articulação ou articulação como uma estrutura multicomponente, limitada pela pele sobreposta, apoiada por osso e cartilagem interiores, e agindo através do nervo intrínseco, músculo, tendão, revestimento articular e vaso, por sua vez, implica que qualquer um destes elementos pode ser impactado para produzir displasia, uma ruptura tecidual distinta das malformações causadas por outras desordens genéticas. O desenvolvimento da clonagem e sequenciamento de DNA confirmaria a operação da herança autossômica dominante e a capacidade dos genes alterarem qualquer elemento da estrutura articular para causar o padrão sintomático do DAA.
Os testes genéticos começaram com um foco em um gene “candidato” selecionado por causa dos sintomas do paciente, ilustrados pelos testes do gene da fibrilina-1 para a síndrome de Marfan . Seqüenciando as letras AGCT do DNA do gene, o “texto” dos nucleotídeos, poderia revelar substituições ou “typos” que alteraram o aminoácido da proteína codificada e sua função pretendida. Então veio a nova tecnologia chamada NextGen ou sequenciamento rápido de DNA paralelo, análogo à leitura de todas as páginas de um livro de uma só vez e não de ponta a ponta . Isto permitiu o sequenciamento de grupos de genes (painéis) como os que causam cardiomiopatia ou displasia do tecido conjuntivo ou mesmo todos os 23.000 genes em cada célula humana (nosso genoma) em 2-3 meses . A sequenciação de regiões de genes codificadores de proteínas ou exônicos, chamada sequenciação de exomas inteiros (WES), pode ser encomendada a muitas empresas e na minha prática foi obtida por menos de $200-300 dólares fora do bolso em metade dos meus pacientes (o preço de lista é $9000 e o valor faturado ao seguro é de cerca de $20.000).
Embora a interpretação da média de 12.000 variantes de DNA detalhadas pelo WES continue sendo um problema, diminuído por uma crescente epidemiologia de variantes em populações normais e doentes, a produção de alterações genéticas que contribuem para os sintomas do DAA entre 620 pacientes submetidos ao WES na minha prática é superior a 60%. Além das mais de 80 mutações nos genes do colágeno, incluindo as do colágeno tipo I geralmente associadas à osteogênese imperfeita, o colágeno tipo V frequentemente associado à SED clássica, os colágenos tipo VI, IX e XII inicialmente descritos com miopatias, e o temido colágeno tipo III associado à SED vascular, são mutações recorrentes nos genes que afetam a pele, nervos, músculos, fibrilares e componentes dos vasos que reforçam um conceito de DAA onde a alteração de qualquer componente articular produz o padrão de sinal e sintoma descrito na Tabela 1. Agora o reconhecimento da SED e desordens relacionadas abrangidas pelo conceito de DAA pode reforçar a impressão clínica com evidências objetivas de DNA que permitem o aconselhamento do paciente e da família.
Diagnóstico e terapia das desordens de hipermobilidade/AAD
Alerta para os principais sintomas de dor/fragilidade articular e autonômicos (intestino, coração, ruptura imunológica) podem levar a testes rápidos para hipermobilidade, como mostrado na Figura 1. O sinal do polegar para o antebraço pode preceder toda a escala Beighton, dando um ponto bilateral para dedos pressionados para trás além dos 90 graus na mão, tocando os polegares nos antebraços, hiperextendendo o cotovelo com os braços estendidos, hiperextendendo os joelhos para formar uma curva para trás das pernas, e 1 ponto para a capacidade de tocar as palmas das mãos no chão . Um breve levantamento de sintomas como os da Tabela 1 pode confirmar a suspeita, auxiliado talvez pela inclusão nos exames físicos de algumas das avaliações listadas na parte inferior da Tabela 1. Note que nem todos os pacientes com DAA são altos ou magros, e que uma porção substancial estará acima do peso devido a restrições de atividade por causa da dor e fadiga. Muitos terão pele elástica, examinada puxando as dobras ao redor da mandíbula ou no antebraço, particularmente quando estas são as dobras finas e superficiais da epiderme. A formação de tecido branco ou formação de quelóide em torno de cicatrizes é comum, e a avaliação de Beighton ou outras manobras de hipermobilidade listadas na Tabela 1 podem ser úteis. Uma postura descida com curva do pescoço para a frente, escoliose e lordose pode ser notada junto com os pés chatos ou o pé chato (mais do que dentro) ao caminhar. A força muscular será reduzida em alguns pacientes, juntamente com o mau equilíbrio pela caminhada em tandem, mas o generalista com tempo limitado precisa atender apenas a alguns desses sinais físicos; o encaminhamento estabelecerá o resto.
Os ortopedistas mais importantes serão os ortopedistas familiarizados com a osteoartrose e as alterações da SED como alterações articulares degenerativas, cartilagem deteriorada, bandas plicas e regiões de osso morto. É desejável um ortopedista experiente e conservador que reconheça os maus resultados das cirurgias para “apertar” ou substituir as articulações. Um cardiologista pode documentar POTS usando o teste de mesa inclinada para alterações ortostáticas na frequência cardíaca e pressão arterial, realizando também estudos eletro e ecocardiográficos para excluir dilatação da aorta ou aneurismas. A neurologia pode avaliar enxaquecas comuns ou dores de cabeça diárias, a deformação do Chiari que ocorre com escorregamento e hérnia de tecido cerebral frouxo, a neuropatia devido à constrição dentro das articulações frouxas e as alterações musculares denotadas pela fibromialgia descritiva dos sintomas e destacadas pelas frequentes alterações do gene miopático que está promovendo um novo termo de SED miopática. Alergista familiarizado com a ativação de mastócitos e gastroenterologistas que podem documentar a baixa motilidade intestinal são adições valiosas para a equipe da subespecialidade, e ninguém é mais valioso do que o médico principal para gerenciar as muitas recomendações e medicamentos que podem ser necessários. Embora possa parecer estranho vindo de um geneticista, penso que as avaliações da subespecialidade que levam a uma taxa de terapia superior à avaliação e testes genéticos, mas esta última pode ser útil para desarmar suposições sobre a doença psiquiátrica .
Tratamento implora com um programa de exercícios que pode beneficiar tanto a artrite devido à hipermobilidade articular como a sua disautonomia reflexiva. A atenção à dor articular (dores de crescimento ocorrem à noite) ou lesão/ deformidade esquelética pode instituir a prevenção, tornando o paciente consciente da sua condição (note na Tabela 1 que muitos não reconhecem a sua flexibilidade como anormal). O desgaste e as lesões nas articulações podem ser prevenidos com o apoio de articulações susceptíveis, com cintas para apoio das costas e com a escolha de actividades de protecção das articulações como natação em vez de jogging. Os pacientes devem permanecer ativos com levantamento de peso moderado e outras atividades razoáveis para construir músculos ao redor das articulações, evitando ciclos de inatividade que exacerbam a fadiga crônica ou fibromialgia. Fisioterapia adequada, controle da dor, orientação de exercícios e medicamentos anedóticos como glucosamina/condroitina têm ajudado muitos, mas devem ser evitados relaxantes que tiram a proteção muscular das articulações e injeções de esteróides que mascaram lesões, promovem osteoporose/degeneração e dão apenas alívio temporário.
A base da terapia de disautonomia é a hidratação (8-10 copos de líquido por dia), sal (sem hipertensão labial), e uma dieta rica em proteínas, tudo com o objectivo de aumentar o volume de líquido sanguíneo e melhorar a circulação cerebral. A terapia nutricional adicional pode incluir vitamina C (2 g por dia), vitamina D (>2000 unidades por dia, mais do que as 400-800 em multivitaminas), vitamina B12 (1-2,5 mg por dia). A baixa motilidade intestinal ou gastroparese é ajudada evitando líquidos antes das refeições, refeições menores e mais frequentes, probióticos, preparados enzimáticos e, pelo relato de muitos, glúten inferior e laticínios que são irritantes comuns. Programas de exercícios supino, biofeedback e muitas opções de medicamentos estão disponíveis através de cardiologistas para POTS (por exemplo, beta-bloqueadores, Midodrine, Florinef), através de neurologistas ou especialistas em gestão da dor (por exemplo, Gabapentina, Lyrica), ou através de alergista que podem fornecer protocolos anti-histamínicos para suprimir mastócitos (por exemplo, Zyrtec, Zantac, Cromolyn, Singulair). Vantagens de testes genómicos como o WES que vão além dos genes conhecidos por causar displasia do tecido conjuntivo são descobertas novas como os genes relacionados com os músculos agora conhecidos por causar SED e documentação de descobertas incidentais ou secundárias como as mutações do gene do cancro da mama. A descoberta de genes associados a sintomas mais graves permite aos pacientes conhecer os riscos para a descendência e escolher estratégias de diagnóstico genético pré-natal ou pré-implantatório compatíveis com seus valores.
Apresentação do caso
Vinha veio ao médico aos 18 anos, tonta, desmaiada, ansiosa e doente do estômago, tão exausta que ela se aposentou às sextas-feiras à noite para descansar todo o fim de semana. Ela tinha sido saudável, activa na ginástica e na alegria, uma rapariga corajosa e brilhante cuja perseverança, apesar das lesões, ganhou os aplausos dos seus treinadores. Depois, uma gripe da qual nunca se recuperou, náuseas tão graves que não conseguia comer, perda de peso, desmaios nas aulas, acordar à noite com o coração a bater, acordar de manhã sem sono reparador. Eva viu muitos médicos, até mesmo as urgências quando as palpitações e o pânico lhe tiraram o fôlego. Nada de errado excepto o stress que eles disseram, tudo na sua mente. Acalmada mas não curada por pílulas, trabalho falhando por cansaço e confusão, ela se perguntava se estava ficando louca. Não se reconhecia sua pele macia e frágil, seus períodos duros e pesados, suas dores de cabeça diárias e, o mais marcante de tudo, a hipermobilidade que ela e sua família supunham ser normal: “Sim, somos benevolentes e com dupla articulação, não são todos?”. Eve teria respondido se perguntado.
Felizmente Eve procurou um novo médico primário que foi capaz de documentar muitos dos sinais e sintomas listados na Tabela I. Eve teve cólicas precoces e refluxo com dificuldade para amamentar, era muito flexível, chamada pela mãe de “bebê sem osso”, e quando mais velha sentava-se de joelhos com as pernas estendidas atrás dela (a posição “W”), dizendo que era mais confortável. Inicialmente ela era lenta a andar, caindo frequentemente e parecendo desajeitada, mas logo estava ativa e subindo por todos os lados, fazendo seus pais tremer, colocando um pé ao redor do pescoço ou fazendo as rachaduras com facilidade. Mas ao entrar na escola primária começou a queixar-se de artralgia, doendo-lhe as pernas à noite com os joelhos e tornozelos a incomodá-la depois da actividade. O pediatra tranquilizou seus pais que ela estava apenas tendo “dores de crescimento”, mas eles ficaram mais preocupados quando ela tentou jogar futebol e continuou virando seus tornozelos, uma entorse exigindo uma chuteira por 2 meses após uma fratura de estresse foi notada.
Eve teve que parar o futebol, mas descobriu que ela se sobressaiu na ginástica à medida que envelhecia, escolhendo exercícios no chão por causa do baixo equilíbrio na viga e uma tendência para o ombro dela puxar para fora nas barras paralelas. Ela notou que as suas articulações estalavam com movimento e muitas vezes estalavam de propósito porque parecia aliviar a tensão e a dor. Ela tentou ser líder de claque com sucesso em manobras, mas descobriu que era demasiado sensível ao calor. As outras raparigas chamavam-lhe “cara de tomate”, pois ela ficava sobreaquecida durante as rotinas, e cansava-se facilmente, precisando muitas vezes de um dia de descanso depois do exercício. Seus problemas digestivos reapareceram com freqüentes constipações e dores de estômago, muitas vezes sentindo-se inchada e precisando de laxantes. No 6º ano ela estava faltando à escola por causa de seus problemas intestinais e dores nas articulações, muitas vezes se sentindo cansada e tonta. Ela não se sentia descansada pela manhã e muitas vezes fazia a sesta aos fins-de-semana, uma saída do seu estilo de vida activo e entusiasta. Muitas vezes ela se sentia ansiosa, não conseguindo acompanhar o estresse escolar ou familiar específico, e começou a ter dores de cabeça pela manhã, o que ela pensava ser devido ao seu sono deficiente. Seus pais a levaram novamente ao pediatra e novamente foram informados de que não havia nada de errado a não ser o habitual estresse escolar.
Quando ela tinha 14 anos, Eve teve uma doença tipo gripe e faltou à escola por um mês com fadiga severa, dores de cabeça, náuseas, vômitos e perda de peso. Ela tinha tonturas terríveis e algumas dores no peito, sentindo o coração bater às vezes (tendo palpitações). Seus pais procuraram outro pediatra e ela mandou Eve a um cardiologista, que achava que ela estava tendo taquicardia devido ao estresse e ansiedade em torno de sua doença. Ele deu a Eve propranolol, um beta-bloqueador, para diminuir o ritmo cardíaco, mas ela ficou ainda mais fatigada e agora estava acamada a maior parte do dia. A dor nas articulações aumentava depois da doença e ela ficava na cama a maior parte do dia, sentindo-se triste e ansiosa quando seus pais tentavam levá-la para fora ou levá-la para dar uma volta. Eles a trouxeram de volta ao segundo pediatra que a encaminhou à psiquiatria, e Zoloft começou a ajudar na ansiedade e na depressão. Depois de alguns dias, Eve teve uma erupção cutânea com urticária e teve que parar a medicação. Ela continuou com tonturas e foi vista por mais dois especialistas, um gastroenterologista que achava que ela poderia ter doença celíaca e um otorrinolaringologista que diagnosticou vertigens e a mandou para a neurologia. Ela contou a esse médico sobre dores de cabeça que emanavam da parte de trás da cabeça, e foi obtido um estudo de ressonância magnética da cabeça supina que era normal.
Apreciação do provável diagnóstico de AAD pelo seu médico primário instituiu uma série diferente de referências de subespecialidade, incluindo neurologia, onde um estudo de ressonância magnética da cabeça ereta demonstrou a hérnia de deformação de Chiari. Um cardiologista documentou um teste de inclinação anormal juntamente com baixa motilidade intestinal e provou que sua ansiedade e fadiga ao estresse eram devidas à disautonomia. Estratégias nutricionais de hidratação, sal e proteína alta com beta-bloqueador e terapia anti-histamínica melhoraram sua fadiga e sintomas intestinais e diminuíram suas dores de cabeça. Foi decidido que a hérnia de Chiari de 4-5 mm não era suficiente para requerer cirurgia, e seu manejo psiquiátrico foi reconhecido como tratando sintomas secundários de um distúrbio médico. Alguns dos seus subspecialistas, cépticos quanto ao seu diagnóstico clínico, foram convencidos por testes genéticos que documentaram uma alteração num gene da cadeia alfa-1 do colagénio tipo V, abreviado COL5A1, um achado comum em pacientes com SED. Eve tinha herdado a mutação de sua mãe, que tinha tido sintomas mais leves de AAD e agora era capaz de lidar melhor com eles.
Durante vários meses a saúde e vitalidade de Eve foram restauradas e ela “recuperou sua vida”, como ela e seus pais diriam. Ela ainda tinha dores nas articulações, ansiedade e fadiga em algumas ocasiões, mas “exercícios sensatos” para construir proteção muscular em torno de suas articulações, abordagens nutricionais para disautonomia e abordagens de biofeedback para afastar a ansiedade mudaram a vida. Esses tratamentos foram todos por causa do reconhecimento por seu médico de família, agora capaz de coordenar os cuidados para Eve e sua mãe e de fornecer recursos para a compreensão tanto da SED quanto das alterações genéticas subjacentes. A transição para a geração seguinte foi a capacidade de informar Eva sobre seus 50% de risco para transmitir sua mutação COL5A e alguns riscos de trabalho de parto precoce e sangramento pós-parto em qualquer gravidez futura .
- Remvig L, Jensen DV, Ward RC (2007) Epidemiologia da hipermobilidade articular geral e base para os critérios propostos para a síndrome de hipermobilidade articular benigna: revisão da literatura. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. In: Tecido conjuntivo e seus distúrbios hereditários. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Distúrbios hereditários do tecido conjuntivo. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Parte C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomia na síndrome de hipermobilidade articular. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lições de alívio da dor – uma experiência pessoal de pós-graduação. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) Uma pequena molécula para uma grande doença. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Capítulo 11: Gene e sequenciação do genoma: Interpretando a variação genética ao nível do nucleotídeo. In: Variação do Cromossoma Humano: Heteromorfismo, Polimorfismo e Patogénese, Ed.2. Springer Nature, Singapura
- Mefford HC (2012) Sequenciação diagnóstica do exoma… Já lá estamos? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Reescrevendo a história natural da dor e sintomas relacionados na síndrome de hipermobilidade articular/Síndrome dehlers-Danlos, tipo hipermobilidade. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Benign joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) A síndrome de hipermobilidade. Queixas músculo-esqueléticas associadas à hipermobilidade articular generalizada. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Diagnóstico genético pré-sintomático e pré-implantação: Neurologia, Próxima Genética, e a próxima geração. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Gestão da dor e da fadiga na síndrome da hipermobilidade articular (também conhecida como síndrome de Ehlers- Danlos, tipo hipermobilidade): princípios e proposta para uma abordagem multidisciplinar. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Reescrevendo a história natural da dor e sintomas relacionados na síndrome da hipermobilidade articular/Síndrome de Ehlers-Danlos, tipo hipermobilidade. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Tratamento multidisciplinar da incapacidade na síndrome de Ehlers-Danlos tipo hipermobilidade/síndrome de hipermobilidade: um estudo piloto utilizando uma combinação de terapia física e cognitiva-comportamental em 12 mulheres. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exome analysis of connective tissue dysplasia: death and renasth of clinical genetics? Am J Med Genet A 164A: 1209-1212.