Nøgleord
hypermobilitet, bindevævsdysplasi, ehlers-danlos syndrom, beighton-skala, dysautonomi, posturalt ortostatisk takykardiesyndrom, irritabel tarmsyndrom, mastcelleaktiveringsforstyrrelse, articulo-autonomisk dysplasi
Indledning
De 20 % af kvinderne og 10 % af mændene, hvis væv er mere fleksibelt og skrøbeligt end gennemsnittet, er meget undervurderet og tragisk dårligt behandlet. Der er få muligheder for forebyggelse og behandling, der er større end at genkende hypermobilitet, som let screenes ved at bede patienten om at presse tommelfingeren mod underarmen (figur 1). Dette afslørende tegn kan vise vejen til en mere detaljeret evaluering, som ligger godt inden for den praktiserende læges område, idet deres brede kliniske viden passer fint til de multisystemproblemer, der kan opstå som følge af slapt bindevæv. Her gennemgår jeg karakteren af hypermobilitetsforstyrrelser, viser, at generel snarere end subspecialiseret viden kan føre til livsændrende behandlinger for disse patienter, og viser, hvordan genetisk testning bringer symptomatiske beskrivelser som fibromyalgi eller kronisk træthedssyndrom ind i præcisionsmedicinens skarpsindige lys.
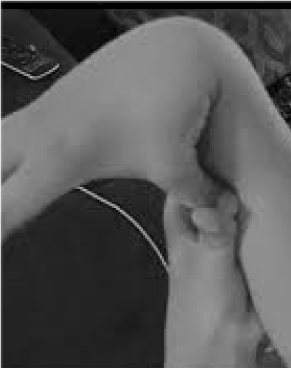
Figur 1. Hypermobilitet.
Der er flere barrierer, der skal overvindes, før hypermobilitetspatienter kan høste fordelene ved almen medicin. Den første stammer indebærer, at dermatologerne Ehlers og Danlos’ banebrydende, men nærsynede fokus har præget utrolige og eksotiske hudstrækninger som det misvisende symbol på bindevævsløshedssygdomme. En anden hindring er den voksende kløft mellem praktisk og genetisk medicin, idet førstnævnte på passende vis koncentrerer sig om almindelige sygdomme, mens sidstnævnte indtil for nylig anvendte specialiseret teknologi til at beskrive de sjældne. De obskure eponymer med skræmmende undertyper og tekniske termer er som små provinser i et uindbydende fremmed land, som ingen global rejsende vil tage sig tid til at kortlægge. For at imødegå disse hindringer er der brug for en praktisk tilgang til hypermobilitetsforstyrrelser, som fokuserer på anerkendelse ved hjælp af anamnese og fysisk undersøgelse og overlader de tekniske detaljer vedrørende eponymer, undertyper og genetisk testning til subspecialister, som kontaktes efter det vigtige første skridt, nemlig den formodede diagnose. Når først den fleksible patient er anerkendt og henvist, forbliver den fleksible patient afhængig af sin primære læge, for kun denne kan integrere anbefalinger og testresultater i det generelle medicinske hjem.
Terminologi
Indskrænket bevægelsesomfang på flere artikulationssteder (led) forekommer i et stort antal sygdomme, lige fra kromosomforstyrrelser som Downs syndrom til sygdomme som Ehlers-Danlos syndrom (EDS), der er kendt for deres hypermobilitet. McKusick populariserede denne kategori af sygdomme, der er kendt for vævsløshed, ved at gruppere dem som arvelige sygdomme i bindevævet, herunder Marfan-syndromet, som han fokuserede på som kardiolog. I 1977 organiserede Dr. Peter Beighton, der er kendt for sin skala til måling af fleksibilitet, den mellemliggende litteratur ved at postulere 6-7 EDS-typer, der nu i de seneste retningslinjer er forenklet som EDS klassisk, vaskulær og hypermobil . Det er nu klart, at disse typer har betydelige overlapninger af symptomer mellem hinanden og beslægtede sygdomme som osteogenesis imperfect (med mange frakturer), Marfan syndrom (stor karudvidelse/dissektion), Stickler syndrom (øjen- og gigtforandringer) og mange andre.
Snarere end at beskæftige mig med bestemte sygdomme finder jeg det mere nyttigt at fokusere på to store sygdomsprocesser fra bindevævsdysplasier, illustreret af de mange symptomer, der er anført i tabel 1. For det første er der de slidte ledsmerter (slidgigt), der adskiller sig fra de hævede, røde led i inflammatorisk (autoimmun eller inflammatorisk) gigt, som man støder på ved reumatoid arthritis, lupus, Sjögrens syndrom eller ankyloserende spondylitis. For det andet er den ubalance i det autonome nervesystem (dysautonomi) forårsaget af elastiske blodkar i hypermobilitetssygdomme . Udspilede kar forårsager blodansamling i bækkenet og benene med lavere blodtryk og svimmelhed/besvimelse ved stående (ortostatisk hypotension). Med tiden aktiverer den nedsatte blodgennemstrømning til hovedet den sympatiske del af det autonome nervesystem (adrenalindelen “flugt eller kamp”-delen), hvilket fører til flere problemer: 1) posturalt ortostatisk takykardiesyndrom (POTS) med takykardi, angst, kronisk træthed og “hjernetåge” (intervaller med nedsat fokus og hukommelse), 2) irritabel tarmsyndrom på grund af hæmning af tarmmotiliteten (parasympatisk undertrykkelse) med forstoppelse vekslende med diarré, oppustethed, refluks, mavesmerter, dysfagi, kvalme og 3) mastcelleaktiveringsforstyrrelse med forbigående udslæt, nældefeber/reaktiv hud, astma/ åndenød og fødevare-/medicinintolerancer. Jeg har udtænkt et bredere begreb, der letter anerkendelsen af enhver hypermobilitetsforstyrrelse, idet jeg anerkender de to vigtigste konsekvenser af led/vævsløshed: articulo-autonomisk dysplasi eller led-fatigue-kompleks.
Tabel 1. Typiske fund og deres hyppighed i hypermobilitetssyndromer. *Fra 320 kvindelige og 54 mandlige patienter over 12 år vurderet ved hjælp af detaljerede spørgeskemaer i 2017; TMJ, temporomandibulært led; POTS, posturalt ortostatisk tachykardisyndrom. Symptomliste delvist tilpasset fra Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
HISTORIE (procenter)*
Vågenhed: Kolik (28), ernæringsvanskeligheder (19,4); klodset, mange fald (29,1)
Barn/teenager: Langsom vægtøgning (12); opmærksom på fleksibilitet (55,6); viste leddetricks (53,6)
tidlige ledsmerter (42,1); teenageaktiviteter begrænset af smerter-skader (55,4); tidlige briller (46,8)
Ordodonti (61,4%) tandkødssygdomme (11,3); dårlig emalje/mange huller (31,6)
Ledsagerproblemer: Popping (81,6); subluxationer (62); TMJ popping, catching, smerter (51,2)
smerter i mindst 2 led (80,3); ledoperationer (33); mange forstuvninger (26); frakturer >2 (31,5)
diskusprolaps i rygmarven (29,3)
Skeletale problemer: Skoliose (31); pectus (2,2); gang-tåning ind/ud (9,3); platfødder (15,5)
Hud: Let blå mærker (61); strækbar hud (15); fløjlsagtig (18); usædvanlige ar (36,3)
striae før graviditet (73); langsom heling (32,4)
Genitourinær (kvinder): (kvinder): Menoragi (61,2); endometriose (11,3); ovariecyster (55)
polycystisk ovariesyndrom (8,5); blæreproblemer (46,1); inguinal/femoral brok (11,5)
Neuromuskulært: Migræne (51,7); daglig hovedpine (62,3); behov for hovedpine-medicin (45)
Chiari-deformation (10,6); neurokirurgi (2,9); følelsesløshed/prikken (48,7); dårlig balance (48,6)
neuropatiske stikkende smerter (37,3); muskelsmerter/spasmer (51,7); muskelsvaghed (32,4)
GI/armhuler: Forstoppelse-diarré (67,1); oppustethed-reflux-mavesmerter (57,3)
Galdeblæreproblemer (12,9); synkebesvær (23); hyppig kvalme (44,9)
Hjerte: Hjerte: Mitralklapprolaps (8,7); aneurisme (0,9); anden arytmi end takykardi (6,8)
POTS: Svimmel ved at stå (69,8); synkope (34,5); kronisk træthed (81,8); søvnproblemer (43,5)
hjernetåge (dårlig hukommelse eller fokus til tider-70,8); varme-/kuldefølsomhed (77,5); unormal svedtendens (25); takykardi (72,8); angst-panikanfald (61,3); lyst til salt (51,2)
Immun/mastcelle: Forbigående udslæt (33,4 %); nældefeber/reaktiv hud (45,9 %); astma/ åndenød (39,3); intolerance over for mad-medikamenter (61,5 %)
Laboratorie/afbildning: Lav knogletæthed (2,1); lavt D-vitamin (28,7); lavt B12-vitamin (2,7); hypothyroid (11,5); lavt ferritin (1,7); lavt jernindhold (6,7); forhøjet anti-nukleært antistof (7,7); forhøjet anti-nukleært antistof (7,7).1)
Gennemsnitligt antal fund ud af 80: kvinder >12 (35,4 fund) mænd >12 (21,5 fund)
Fysisk (procent)
Bygning: Høj højde >90. percentil (55,2); tung-BMI >28 (21,3); tynd-BMI <19 (14,2)
Marfanoid habitus (44); arachnodactyly (46,4); Walker-Murdoch tegn til stede (40,2)
Face: Lang (25,7); stramt underansigt (5,4); blågrå sclera (1,6); høj gane (53,5)
Hud: Blød (81,4); gennemskinnelig (23,8); elastisk-lift >1 tommer fold omkring kæben på midten af underarmen (51,3)
Folder består af ydre (epidermisk) lag (35.4); usædvanlige ar (42,1 %)
Beighton-score: 0-3 point (8,4); 4-6 point (32,6); 7-8 point (38,8); 7-8 point (38,8); 9 af 9 point (20,2)
Andre manøvrer: Saml hænderne over skulder- bag ryggen (68)
opadgående bønnetegn bag ryggen (71,3); før hånden rundt om ryggen for at berøre navlen (19,5)
Skelet: Nakke kyfose (41,2); skoliose (22,3); lordose (32,8); flade fødder (39,6)
gangart – at gå indad eller udad (28,3)
Neurologisk: Neurologisk: Muskelsvaghed (11,2); nedsat muskelmasse (8,8)
dårlig balance ved tandemgang (33,4)
Gennemsnitligt antal fund ud af 40: kvinder >12 (17,9 fund) mænd >12 (15.7 fund)
Articulo-autonomisk dysplasi (AAD)
Et begreb som “articulo-autonomisk dysplasi” eller AAD kan forene tilgangen til bindevævslaksitet/hypermobilitetsforstyrrelser og imødegå tre problemer, der vanskeliggør deres genkendelse: 1) mange læger og genetikere betragter bindevævsdysplasier som sjældne og ekstreme sygdomme, når de i virkeligheden rammer ca. 1 % af befolkningen 2) diagnostiske retningslinjer for EDS, selv om de forenkler de vigtigste EDS-typer som klassisk, vaskulær og hypermobilitet, forvirrer stadig ved at henvise til mange mindre typer, der udgør et overlappende symptomspektrum, og 3) den næsten obligatoriske dysautonomi, der ledsager EDS og beslægtede lidelser (tabel 1), udelades som et diagnostisk kriterium, svarende til at beskrive en elefant uden at nævne dens snabel. AAD betegner den uundgåelige fysiologiske forbindelse mellem løst væv og dysautonomi på samme måde som de metaboliske eller hepatorenale syndromer beskriver, hvordan fedme fører til leversygdom/diabetes eller leversvigt forårsager nyresygdom.
Som de fleste medicinske mønstre eller syndromer, herunder EDS, er AAD et symptomkompleks med mange underliggende årsager. “Articulo” henviser til leddet, idet man erkender, at lidelse af enhver ledkomponent – hud, nerve, muskel, knogle, bindevæv, blodkar – forårsager et lignende mønster af ledvævets slaphed med autonom ubalance. Der vil være skrøbelighed i huden, nedsat knogletæthed med risiko for brud, muskelsvaghed med smerter (myalgi), neuropati med følelsesløshed/prikken, karpaltunnel eller regionale smertesyndromer, autonome ændringer, herunder takykardi, angst, ikke-restaurativ søvn, tarmimmotilitet, nældefeber/udslæt, fødevare- og medicinintolerans, som alle er til stede i varierende omfang selv inden for specifikke “typer” af EDS. Jeg har ikke fundet store forskelle mellem patienter, der passer til bestemte typer EDS, hvilket illustreres af hyppigere hudforandringer i type I klassisk cEDS versus større fleksibilitet i leddene i type III hypermobil hEDS. Hyppigheden af symptomer hos 374 patienter registreret ved systematisk forespørgsel varierede ikke væsentligt blandt dem, jeg diagnosticerede som cEDS versus hEDS, og selv de 8 patienter, der blev identificeret som mulige vaskulære vEDS ved at have kollagentype III-genmutationer, var ikke særpræget (aneurismer, der var til stede hos en af de 8 patienter, havde samme lave hyppighed i andre EDS-kategorier). Det vigtigste er konceptet om et EDS-spektrum, der efter min mening bedst repræsenteres som AAD.
Erkendelse af AAD-symptomplekset, der skyldes EDS og beslægtede bindevævsdysplasier, vil forhindre triste patientrejser fra specialist til specialist, symptomatiske diagnoser som svimmelhed, besvimelse, fibromyalgi eller kronisk træthed og tragisk lidelse, når mange forebyggende strategier og behandlinger er til rådighed. Desuden forklarer anerkendelse af dysautonomi angsten ved POTS og den depression, der ofte ledsager kroniske smerter eller aktivitetsbegrænsning , adfærdsforskelle, der kan anerkendes som konsekvenser af medicinsk snarere end psykiatrisk sygdom.
Genetisk testning understøtter konceptet om articulo-autonomisk dysplasi
Implementeret af deres karakterisering som arvelige lidelser af bindevæv , blev de mere ekstreme former for EDS og beslægtede lidelser fundet at udvise autosomal dominant arv, hvilket betyder, at de berørte individer har en normal og en unormal form af genet (allel). Dominant arv giver mening, fordi det defekte protein, der produceres af den unormale allel, svarende til en skæv mursten, ville blande sig med normale mursten fra den anden allel og skabe en vakkelvorn struktur. Anerkendelse af leddet eller leddet som en struktur med flere komponenter, der er begrænset af den overliggende hud, understøttet af indvendig knogle og brusk og virker gennem indre nerve, muskel, sene, ledforing og kar, indebærer igen, at ethvert af disse elementer kan påvirkes og give dysplasi, en vævsforstyrrelse, der adskiller sig fra de misdannelser, der forårsages af andre genetiske sygdomme. Udviklingen af DNA-kloning og sekventering vil kunne bekræfte, at der er tale om autosomal dominerende arv og at gener, der ændrer et hvilket som helst element i ledstrukturen, kan forårsage AAD’s symptommønster.
Genetisk testning begyndte med fokus på et “kandidatgen”, der blev udvalgt på grund af patientens symptomer, hvilket blev illustreret af fibrillin-1-gen-testning for Marfans syndrom . Ved at sekventere genets DNA AGCT-bogstaver, “teksten” af nukleotider, kunne man afsløre substitutioner eller “skrivefejl”, som ændrede aminosyren i det kodede protein og dets tilsigtede funktion. Så kom den nye teknologi kaldet NextGen eller hurtig parallel DNA-sekventering, som svarer til at læse alle siderne i en bog på én gang i stedet for fra ende til anden . Dette gjorde det muligt at sekventere gengrupper (paneler) som f.eks. dem, der forårsager kardiomyopati eller bindevævsdysplasi, eller endog alle 23 000 gener i hver enkelt menneskelig celle (vores genom) på 2-3 måneder . Sekventering af de proteinkodende eller exoniske genregioner, kaldet whole exome sequencing (WES), kan bestilles hos mange firmaer og blev i min praksis opnået for mindre end 200-300 USD ud af egen lomme hos halvdelen af mine patienter (listeprisen er 9000 USD og det beløb, der faktureres til forsikringen, er omkring 20 000 USD).
Og selv om fortolkningen af de gennemsnitligt 12.000 DNA-varianter, der er detaljeret beskrevet ved WES, fortsat er et problem, der mindskes af en voksende epidemiologi af varianter i normale og sygdomspopulationer, er udbyttet af genændringer, der bidrager til AAD-symptomer blandt 620 patienter, der gennemgår WES i min praksis, over 60 %. Ud over over 80 mutationer i kollagengener, herunder dem i kollagen type I, der normalt er forbundet med osteogenesis imperfecta, kollagen type V, der ofte er forbundet med klassisk EDS, kollagen type VI, IX og XII, der oprindeligt blev beskrevet med myopatier, og det frygtede kollagen type III, der er forbundet med vaskulær EDS, er der tilbagevendende mutationer i gener, der påvirker hud-, nerve-, muskel-, fibrillær- og karkomponenter, der styrker et koncept af AAD, hvor ændring af en hvilken som helst ledkomponent producerer det tegn- og symptommønster, der er skitseret i tabel 1. Nu kan genkendelse af EDS og beslægtede lidelser, der er omfattet af AAD-konceptet, underbygge det kliniske indtryk med objektive DNA-beviser, der muliggør rådgivning af patient og familie.
Diagnostik og behandling af hypermobilitetsforstyrrelser/AAD
Opmærksomhed på de vigtigste symptomer på ledsmerter/fragilitet og autonome (tarm, hjerte, immunforstyrrelser) kan føre til hurtige tests for hypermobilitet som vist i figur 1. Tommelfinger til underarmstegnet kan indlede hele Beighton-skalaen og give et point bilateralt for fingre presset tilbage ud over 90 grader på hånden, berøring af tommelfingre til underarme, hyperextension af albuen med strakte arme, hyperextension af knæene for at danne en bagudvendt benkurve og 1 point for evnen til at røre håndfladerne mod gulvet . En kort undersøgelse af symptomer som dem i tabel 1 kan bekræfte mistanken, eventuelt ved at lade nogle af de vurderinger, der er anført i den nederste del af tabel 1, indgå i de fysiske undersøgelser. Bemærk, at ikke alle AAD-patienter er høje eller tynde, og at en betydelig del af dem vil være overvægtige på grund af aktivitetsbegrænsninger på grund af smerter og træthed. Mange vil have elastisk hud, som undersøges ved at trække folder op omkring kæben eller på underarmen, især når der er tale om de tynde, overfladiske folder af epidermis. Opbygning af hvidt væv eller keloiddannelse omkring ar er almindelig, og vurdering af Beighton- eller andre hypermobilitetsmanøvrer, der er anført i tabel 1, kan være nyttige. Der kan konstateres en nedsunken kropsholdning med fremadrettet nakkekurve, skoliose og lordose sammen med flade fødder eller udadvendte tæer (mere end indadvendte) ved gang. Muskelstyrken vil være nedsat hos nogle patienter sammen med dårlig balance ved tandemgang, men en generalist med begrænset tid behøver kun at tage sig af nogle få af disse fysiske tegn; resten vil blive fastslået ved henvisning.
Det vigtigste vil være ortopædkirurger, der er bekendt med slidgigt og ledsageforandringer i EDS som degenerative ledforandringer, nedbrydende brusk, plica-bånd og områder med død knogle. Det er ønskeligt med en erfaren og konservativ ortopædkirurg, der erkender de dårlige resultater af operationer for at “stramme” eller udskifte led, er ønskeligt. En kardiolog kan dokumentere POTS ved hjælp af tilt-table-test for ortostatiske ændringer i hjertefrekvens og blodtryk og kan også udføre elektro- og ekkokardiografiske undersøgelser for at udelukke aortaudvidelse eller aneurismer. Neurologi kan vurdere almindelig migræne eller daglig hovedpine, Chiari-deformationen, der opstår ved glidning og herniation af slapt hjernevæv, neuropati som følge af indsnævring i slappe led og muskelforandringer, der betegnes ved symptombeskrivelsen fibromyalgi og fremhæves ved hyppige myopatiske genforandringer, der fremmer en ny betegnelse for myopatisk EDS . Allergologer, der er bekendt med mastcelleaktivering, og gastroenterologer, der kan dokumentere lav tarmmotilitet, er værdifulde tilføjelser til det subspecialiserede team, og ingen er mere værdifulde end den primære læge til at håndtere de mange anbefalinger og den medicinering, der kan være nødvendig. Selv om det kan virke mærkeligt at komme fra en genetiker, mener jeg, at de subspecialitetsvurderinger, der fører til terapi, vurderes højere end genetisk evaluering og testning, men sidstnævnte kan være nyttige til at afvæbne antagelser om psykiatrisk sygdom .
Behandlingen begynder med et træningsprogram, der kan gavne både gigt på grund af ledhypermobilitet og dens refleksive dysautonomi. Opmærksomhed på ledsmerter (voksende smerter opstår om natten) eller skader/skeletdeformitet kan indstifte forebyggelse ved at gøre patienten opmærksom på sin tilstand (bemærk i tabel 1, at mange ikke genkender deres fleksibilitet som unormal). Slidskader kan forebygges ved at afstive modtagelige led, taljebånd til støtte for ryggen og ved at vælge ledbeskyttende aktiviteter som svømning frem for jogging. Patienterne bør forblive aktive med moderat vægtløftning og andre fornuftige aktiviteter for at opbygge muskler omkring leddene og forhindre cyklusser af inaktivitet, der forværrer kronisk træthed eller fibromyalgi. Passende fysioterapi, smertebehandling, træningsvejledning og anekdotisk medicin som glucosamin/chondroitinpræparater har hjulpet mange, men afslappende midler, der fjerner muskelbeskyttelsen af leddene, og steroidinjektioner, der maskerer skader, fremmer osteoporose/degeneration og kun giver midlertidig lindring, bør undgås.
Hovedhjørnestenen i behandlingen af dysautonomi er hydrering (8-10 glas væske om dagen), salt (fraværende labilt hypertension) og en proteinrig kost, der alle har til formål at øge blodets væskevolumen og forbedre hjernecirkulationen. Yderligere ernæringsbehandling kan omfatte C-vitamin (2 g pr. dag) D-vitamin (>2000 enheder pr. dag, mere end de 400-800 i multivitaminer), B12-vitamin (1-2,5 mg pr. dag). Lav tarmmotilitet eller gastroparese hjælpes ved at undgå væske før måltider, mindre og hyppigere måltider, probiotika, enzympræparater og, ifølge mange meldinger, mindre gluten og mejeriprodukter, som er almindelige irritanter. Der findes træningsprogrammer på ryggen, biofeedback og mange medicinske muligheder gennem kardiologer for POTS (f.eks. betablokkere, Midodrine, Florinef), gennem neurologer eller smertebehandlingseksperter (f.eks. Gabapentin, Lyrica) eller gennem allergologer, der kan tilbyde antihistaminprotokoller til at undertrykke mastceller (f.eks. Zyrtec, Zantac, Cromolyn, Singulair). Fordelene ved genomisk testning som WES, der går ud over de gener, der er kendt for at forårsage bindevævsdysplasi, er nye fund som f.eks. de muskelrelaterede gener, der nu er kendt for at forårsage EDS, og dokumentation af tilfældige eller sekundære fund som f.eks. mutationer i bryst- og æggestokkræftgenet. Ved at finde frem til gener, der er forbundet med mere alvorlige symptomer, kan patienterne kende risikoen for deres afkom og vælge strategier for prænatal eller præimplantationsgenetisk diagnose, der er forenelige med deres værdier.
Sagpræsentation
Eve kom til sin læge i en alder af 18 år, svimmel, svag, ængstelig og dårlig i maven, så udmattet, at hun trak sig tilbage fredag aften for at hvile hele weekenden. Hun havde været sund og rask, aktiv i gymnastik og cheerleader, en modig og kvik pige, hvis udholdenhed på trods af skader gav hendes trænere rosende ord med på vejen. Så kom influenzaen, som hun aldrig kom sig over, kvalme så voldsom, at hun ikke kunne spise, vægttab, besvimelse i klassen, opvågnen om natten med et bankende hjerte, opvågnen om morgenen uden genoprettende søvn. Eve opsøgte mange læger, selv skadestuen, da hjertebanken og panik tog pusten fra hende. De sagde, at der ikke var noget galt, bortset fra stress, og at det hele var i hendes sind. Hun blev beroliget, men ikke helbredt af piller, og da hun ikke kunne arbejde på grund af træthed og forvirring, spekulerede hun på, om hun var ved at blive skør. Ukendt var hendes bløde og skrøbelige hud, hendes hårde og kraftige menstruation, hendes daglige hovedpine og, mest slående af alt, hypermobilitet, som hun og hendes familie antog var normalt: “Ja, vi er bøjelige og dobbeltleddet, er alle ikke det?” Eve ville have svaret, hvis hun var blevet spurgt.
Formiddag opsøgte Eve en ny primær læge, som var i stand til at dokumentere mange af de tegn og symptomer, der er anført i tabel I. Eve havde tidlig kolik og refluks med problemer med at amme, var meget fleksibel, blev af sin mor kaldt den “benløse” baby, og da hun blev ældre, ville hun sidde på sine knæ med benene strakt bag sig (W-stillingen), idet hun sagde, at det var mere behageligt. I begyndelsen var hun langsom til at gå, faldt ofte ned og virkede klodset, men snart var hun aktiv og klatrede overalt, og hun fik sine forældre til at trække vejret ved at lægge en fod om halsen på hende eller lave spagat med lethed. Men da hun kom i folkeskolen, begyndte hun at klage over arthralgier, idet hun fik ondt i benene om natten, og hendes knæ og ankler generede hende efter aktivitet. Hendes børnelæge forsikrede hendes forældre om, at hun bare havde “vækstsmerter”, men de blev mere bekymrede, da hun forsøgte at spille fodbold og blev ved med at vende sine ankler om, og en forstuvning krævede en støvle i to måneder, efter at der var konstateret et stressbrud.
Eve måtte stoppe med fodbold, men fandt ud af, at hun udmærkede sig ved gymnastik, da hun blev ældre, idet hun valgte gulvøvelser på grund af dårlig balance på bjælken og en tendens til, at hendes skulder trak ud på de parallelle stænger. Hun lagde mærke til, at hendes led sprang i forbindelse med bevægelse og sprang ofte med vilje, fordi det syntes at lindre spændinger og smerter. Hun prøvede at være cheerleader med succes i manøvrer, men fandt ud af, at hun var for følsom over for varme. De andre piger kaldte hende “tomatansigt”, da hun blev overophedet under rutinerne, og hun blev let træt og havde ofte brug for en hviledag efter træning. Hendes fordøjelsesproblemer dukkede op igen med hyppig forstoppelse og mavesmerter, og hun følte sig ofte oppustet og havde brug for afføringsmidler. I 6. klasse manglede hun skole på grund af sine tarmproblemer og ledsmerter, og hun følte sig ofte træt og svimmel. Hun følte sig ikke udhvilet om morgenen og tog ofte en lur i weekenden, hvilket var en afvigelse fra hendes tidligere aktive og entusiastiske livsstil. Hun følte sig ofte ængstelig, uden at kunne spore det til nogen specifik skole- eller familiestress, og hun begyndte at få hovedpine om morgenen, som hun troede skyldtes hendes dårlige søvn. Hendes forældre tog hende igen med til børnelægen og fik igen at vide, at der ikke var noget galt, men at der var sædvanlig skolestress.
Da hun var 14 år gammel, fik Eve en slem influenzalignende sygdom og udeblev fra skole i en måned med alvorlig træthed, hovedpine, kvalme, opkastninger og vægttab. Hun havde frygtelig svimmelhed og nogle smerter i brystet og følte, at hendes hjerte bankede til tider (havde hjertebanken). Hendes forældre søgte råd hos en anden børnelæge, og hun sendte Eve til en kardiolog, som mente, at hun havde takykardi på grund af stress og angst i forbindelse med hendes sygdom. Han gav Eve propranolol, en betablokkere, for at sænke hendes hjerterytme, men hun blev endnu mere træt og var nu sengeliggende en stor del af dagen. Hendes ledsmerter tog til efter hendes sygdom, og hun blev liggende i sengen det meste af dagen og følte sig trist og blev ængstelig, når hendes forældre forsøgte at tage hende med ud eller køre en tur med hende. De bragte hende tilbage til den anden børnelæge, som henviste hende til psykiatrien, og man begyndte at give hende Zoloft for at hjælpe hende med angst og depression. Efter få dage fik Eve udslæt med nældefeber og måtte stoppe med medicinen. Hun blev ved med at have svimmelhed og blev tilset af yderligere to specialister, en gastroenterolog, som mente, at hun måske havde cøliaki, og en øre-næse-hals-specialist, som diagnosticerede svimmelhed og sendte hende til neurologi. Hun fortalte denne læge om hovedpine, der udgik fra hendes baghoved, og der blev foretaget en MRI-undersøgelse af hovedet i rygliggende stilling, som var normal.
Hendes primære læges vurdering af den sandsynlige AAD-diagnose indledte en anden runde af subspecialiserede henvisninger, herunder neurologi, hvor en MRI-undersøgelse af hovedet i opretstående stilling påviste herniation af Chiari-deformationen. En kardiolog dokumenterede en unormal tilt-table-test sammen med lav tarmmotilitet og beviste, at hendes angst og stress-fatigue skyldtes dysautonomi. Ernæringsmæssige strategier med hydrering, salt og højt proteinindhold samt betablokker- og antihistaminbehandling forbedrede hendes træthed og tarmsymptomer og mindskede hendes hovedpine. Det blev besluttet, at Chiari-herniationen på 4-5 mm ikke var tilstrækkelig til at kræve operation, og hendes psykiatriske behandling blev anerkendt som behandling af sekundære symptomer på en medicinsk lidelse. Nogle af hendes subspecialister, der var skeptiske over for hendes kliniske diagnose, blev overbevist af genetiske tests, der dokumenterede en ændring i et gen for alfa-1-kæden af type V kollagen, forkortet COL5A1, et almindeligt fund hos patienter med EDS. Eve havde arvet mutationen fra sin mor, som havde haft mildere AAD-symptomer og nu var i stand til at håndtere dem bedre.
I løbet af flere måneder blev Eves helbred og vitalitet genoprettet, og hun “fik sit liv tilbage”, som hun og hendes forældre ville sige. Hun havde stadig ledsmerter, angst og træthed af og til, men “klog motion” for at opbygge muskelbeskyttelse omkring hendes led, ernæringsmæssige tilgange for dysautonomi og biofeedback-tilgange for at afværge angst var livsforandrende . Disse behandlinger var alle på grund af anerkendelse fra hendes praktiserende læge, som nu var i stand til at koordinere pleje for Eve og hendes mor og til at give ressourcer til at forstå både EDS og de underliggende genændringer . Overgangen til næste generation var evnen til at informere Eve om hendes 50 % risiko for at videregive sin COL5A-mutation og nogle risici for tidlig fødsel og blødning efter fødslen i enhver fremtidig graviditet .
- Remvig L, Jensen DV, Ward RC (2007) Epidemiologi af generel ledhypermobilitet og grundlaget for de foreslåede kriterier for benignt ledhypermobilitetssyndrom: gennemgang af litteraturen. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) Ehlers-Danlos syndromet. In: Bindevæv og dets arvelige lidelser. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomia in the joint hypermobility syndrome. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lessons in pain relief–a personal postgraduate experience. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) Et lille molekyle til en stor sygdom. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Kapitel 11: Gen- og genomsekventering: Fortolkning af genetisk variation på nukleotidniveau. In: Human Chromosome Variation: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing–are we there yet? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Benign joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Muskuloskeletale klager i forbindelse med generaliseret ledhypermobilitet. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Præsymptomatisk og præimplantationsgenetisk diagnose: Neurologi, NextGenetics og den næste generation. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Håndtering af smerter og træthed i forbindelse med hypermobilitetssyndromet (alias Ehlers-Danlos syndrom, hypermobilitetstype): principper og forslag til en tværfaglig tilgang. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Multidisciplinær behandling af handicap ved Ehlers-Danlos syndrom hypermobilitetstype/hypermobilitetssyndrom: en pilotundersøgelse med en kombination af fysisk og kognitiv adfærdsterapi på 12 kvinder. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exome analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am J Med Genet A 164A: 1209-1212.