Keywords
hypermobiliteit, bindweefseldysplasie, ehlers-danlos syndroom, beighton schaal, dysautonomie, posturaal orthostatisch tachycardie syndroom, prikkelbare darm syndroom, mestcel activatie stoornis, articulo-autonomische dysplasie
Inleiding
Veel te weinig herkend en tragisch verkeerd behandeld zijn de 20% van de vrouwen en 10% van de mannen waarvan de weefsels flexibeler en fragieler zijn dan gemiddeld . Weinig mogelijkheden voor preventie en behandeling zijn groter dan die van het herkennen van hypermobiliteit, gemakkelijk te screenen door de patiënt te vragen zijn duim tegen zijn onderarm te drukken (figuur 1). Dit signaal kan de weg wijzen naar een meer gedetailleerde evaluatie die binnen het bereik ligt van de huisarts, wiens brede klinische kennis goed aansluit bij de multisysteemproblemen die kunnen ontstaan door laks bindweefsel. Hier geef ik een overzicht van de aard van hypermobiliteitsstoornissen, laat ik zien dat algemene in plaats van subspecialistische kennis kan leiden tot levensveranderende therapieën voor deze patiënten, en laat ik zien hoe genetische testen symptomatische beschrijvingen als fibromyalgie of chronisch vermoeidheidssyndroom in het onderscheidende licht van de precisiegeneeskunde brengen.
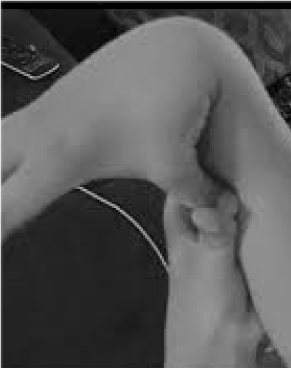
Figuur 1. Hypermobiliteit.
Er moeten verschillende barrières worden overwonnen voordat hypermobiliteitspatiënten de vruchten van de algemene geneeskunde kunnen plukken. De eerste heeft te maken met de baanbrekende maar kortzichtige focus van dermatologen Ehlers en Danlos die ongelooflijke en exotische huidstrekkingen inprenten als het misleidende symbool van bindweefselluxatiestoornissen. Een tweede barrière is de toenemende kloof tussen praktische en genetische geneeskunde, waarbij de eerste zich terecht concentreert op veel voorkomende ziekten, terwijl de laatste tot voor kort gespecialiseerde technologie gebruikte om de zeldzame te beschrijven. De obscure eponiemen met afschrikwekkende subtypes en technische termen zijn als kleine provincies van een ongenaakbaar vreemd land dat geen wereldreiziger de tijd zal nemen om in kaart te brengen. Om deze hindernissen uit de weg te ruimen is een praktische benadering van hypermobiliteitsstoornissen nodig, waarbij de nadruk ligt op herkenning door anamnese en lichamelijk onderzoek, en waarbij de technische details van eponiemen, subtypes en genetische tests worden overgelaten aan subspecialisten met wie na de vitale eerste stap van de vermoedelijke diagnose contact wordt opgenomen. Eenmaal herkend en doorverwezen blijft de flexibele patiënt afhankelijk van zijn huisarts, want alleen die kan aanbevelingen en testresultaten integreren in het algemene medische huis.
Terminologie
Verhoogd bewegingsbereik op meerdere gewrichtsplaatsen komt voor bij een groot aantal ziekten, variërend van chromosoomafwijkingen zoals het syndroom van Down tot aandoeningen zoals het Ehlers-Danlos syndroom (EDS) dat bekend staat om zijn hypermobiliteit. McKusick populariseerde deze categorie van aandoeningen die bekend staan om hun weefsellaxiteit door ze te groeperen als erfelijke aandoeningen van het bindweefsel, waaronder het syndroom van Marfan waar hij zich als cardioloog op richtte. In 1977 organiseerde Dr. Peter Beighton, bekend om zijn meetschaal voor soepelheid, de tussenliggende literatuur door 6-7 EDS types te postuleren, nu vereenvoudigd door recente richtlijnen als EDS klassiek, vasculair, en hypermobiel. Het is nu duidelijk dat deze types een aanzienlijke overlap van symptomen hebben onder elkaar en verwante aandoeningen zoals osteogenesis imperfect (met talrijke fracturen), Marfan syndroom (grote vaatverwijding/dissectie), Stickler syndroom (oog en artritis veranderingen), en vele anderen.
In plaats van me bezig te houden met bepaalde ziekten, vind ik het nuttiger om me te concentreren op twee belangrijke ziekteprocessen van bindweefsel dysplasieën, geïllustreerd door de vele symptomen die in tabel 1 staan. Ten eerste is er de slijtage-pijn van de gewrichten (artrose) die verschilt van de gezwollen, rode gewrichten van inflammatoire (auto-immune of ontstekings) artritis die men aantreft bij reumatoïde artritis, lupus, Sjogren syndroom, of ankylosing spondylitis. Ten tweede is er de verstoring van het autonome zenuwstelsel (dysautonomie) door elastische bloedvaten bij hypermobiliteitsstoornissen. Vernauwde bloedvaten veroorzaken een samenklontering van bloed in het bekken en de benen, met verlaagde bloeddruk en duizeligheid/slapte bij het opstaan (orthostatische hypotensie). Na verloop van tijd activeert de verminderde bloedstroom naar het hoofd de sympathische arm van het autonome zenuwstelsel (de adrenaline “vlucht of vecht” arm), wat leidt tot verschillende problemen: 1) posturaal orthostatisch tachycardie syndroom (POTS) met tachycardie, angst, chronische vermoeidheid en “hersenmist” (intervallen van verminderde focus en geheugen), 2) prikkelbare darm syndroom door remming van de intestinale motiliteit (parasympatische onderdrukking) met constipatie afgewisseld met diarree, opgeblazen gevoel, reflux, maagpijn, dysfagie, misselijkheid, en 3) mestcel activeringsstoornis met voorbijgaande huiduitslag, netelroos/reactieve huid, astma/kortademigheid, en voedsel/medicijn intoleranties. Ik heb een bredere term bedacht die de herkenning van elke hypermobiliteitsstoornis vergemakkelijkt en de twee belangrijkste gevolgen van gewrichts-/weefsellaxiteit erkent: articulo-autonomische dysplasie of gewrichtsmoeheid complex.
Tabel 1. Typische bevindingen en hun frequentie bij hypermobiliteitssyndromen. *Van 320 vrouwelijke en 54 mannelijke patiënten ouder dan 12 jaar beoordeeld met gedetailleerde vragenlijsten in 2017; TMJ, temporomandibulair gewricht; POTS, posturaal orthostatisch tachycardiesyndroom. Symptoomlijst gedeeltelijk aangepast uit Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
HISTORIE (percentages)*
Infanterie: Koliek (28), voedingsproblemen (19,4); onhandig, veel vallen (29,1)
Kind/tiener: Trage gewichtstoename (12); bewust van flexibiliteit (55,6); vertoonde gewrichtstrucs (53,6)
vroeg gewrichtspijn (42,1); tieneractiviteiten beperkt door pijn-blessures (55,4); vroege bril (46,8)
orthodontie (61,4%) tandvleesaandoening (11,3); slecht glazuur/veel gaatjes (31,6)
Gewrichtsproblemen: Popping (81,6); subluxuxaties (62); TMJ popping, catching, pijn (51,2)
pijn in minstens 2 gewrichten (80,3); gewrichtsoperaties (33); veel verstuikingen (26); fracturen >2 (31,5)
wervelschijfherniatie (29,3)
Skeletkwesties: Scoliose (31); pectus (2,2); loop-tenen in/uit (9,3); platvoeten (15,5)
Huid: Gemakkelijk blauwe plekken (61); rekbare huid (15); fluweelachtig (18); ongewone littekens (36,3)
striae voor zwangerschap (73); trage genezing (32,4)
Genitourinair (vrouwen): Menorragie (61,2); endometriose (11,3); cysten in de eierstokken (55)
polycysteus ovarium syndroom (8,5); blaasproblemen (46,1); liesbreuk/femorale hernia (11,5)
Neuromusculair: Migraine (51,7); dagelijkse hoofdpijn (62,3); medicijnen tegen hoofdpijn nodig (45)
Chiari misvorming (10,6); neurochirurgie (2,9); gevoelloosheid/tintelingen (48,7); slecht evenwicht (48,6)
neuropathische pijnscheuten (37,3); spierpijn/krampen (51,7); spierzwakte (32,4)
GI/boezems: Constipatie-diarree (67,1); opgeblazen gevoel-reflux-maagpijn (57,3)
Galblaasproblemen (12,9); moeilijk slikken (23); frequente misselijkheid (44,9)
Hart: Mitralisklepprolaps (8,7); aneurysma (0,9); andere aritmie dan tachycardie (6,8)
POTS: Duizelig bij staan (69,8); syncope (34,5); chronische vermoeidheid (81,8); slaapproblemen (43,5)
hersenmist (soms slecht geheugen of slechte focus-70,8); warmte-/koudegevoeligheid (77,5); abnormaal zweten (25); tachycardie (72,8); angst-paniekaanvallen (61,3); zoutzucht (51,2)
Immuun/mastcel: Voorbijgaande huiduitslag (33,4%); netelroos/reactieve huid (45,9%); astma/kortademigheid (39,3); voedsel-medicatie-intoleranties (61,5%)
Lab/beeldvorming: Lage botdichtheid (2,1); laag vitamine D (28,7); laag vitamine B12 (2,7); hypothyreoïdie (11,5); laag ferritine (1,7); laag ijzergehalte (6,7); verhoogd anti-nucleair antilichaam (7.1)
Gemiddeld aantal bevindingen op 80: vrouwen >12 (35,4 bevindingen) mannen >12 (21,5 bevindingen)
Fysiek (percentage)
Buishouding: Lengte >90e percentiel (55,2); zwaar-BMI >28 (21,3); dun-BMI <19 (14,2)
Marfanoïde habitus (44); arachnodactylie (46,4); Walker-Murdoch-teken aanwezig (40,2)
Gezicht: Lang (25,7); strak ondergezicht (5,4); blauwgrijze sclera (1,6); hoog gehemelte (53,5)
Huid: Zacht (81,4); doorschijnend (23,8); elastisch-lift >1 inch plooi rond kaak op midden-voorarm (51,3)
Plooien bestaan uit buitenste (epidermale) laag (35.4); ongebruikelijke littekens (42,1%)
Beighton-score: 0-3 punten (8,4); 4-6 punten (32,6); 7-8 punten (38,8); 9 van 9 punten (20,2)
Andere manoeuvres: Handen over de schouder achter de rug (68)
omhoog gebedsteken achter de rug (71.3); hand rond de rug brengen om navel aan te raken (19.5)
Skelet: Nekkyfose (41,2); scoliose (22,3); lordose (32,8); platvoeten (39,6)
gang-teen-in- of uittrekken (28,3)
Neurologisch: Spierzwakte (11,2); verminderde spiermassa (8,8)
slecht evenwicht door tandem lopen (33,4)
Gemiddeld aantal bevindingen op 40: vrouwen >12 (17,9 bevindingen) mannen >12 (15.7 bevindingen)
Articulo-Autonomische Dysplasie (AAD)
Een term als “articulo-autonomische dysplasie” of AAD kan de benadering van bindweefsellaxiteit/hypermobiliteitsstoornissen verenigen en drie problemen tegengaan die de herkenning ervan bemoeilijken: 1) veel artsen en genetici beschouwen bindweefsel dysplasieën als zeldzame en extreme ziekten terwijl ze in feite ongeveer 1% van de bevolking treffen 2) diagnostische richtlijnen voor EDS, hoewel ze de belangrijkste EDS types vereenvoudigen als klassiek, vasculair en hypermobiliteit, verwarren nog steeds door te verwijzen naar vele kleinere types die een overlappend symptoom spectrum vormen, en 3) de bijna obligate dysautonomie die gepaard gaat met EDS en verwante aandoeningen (tabel 1) wordt weggelaten als diagnostisch criterium, analoog met het beschrijven van een olifant zonder zijn slurf te vermelden. AAD duidt op het onvermijdelijke fysiologische verband tussen loslippigheid en dysautonomie, op dezelfde manier als de metabole of hepatorenale syndromen beschrijven hoe obesitas leidt tot leverziekte/diabetes of leverfalen nierziekte veroorzaakt.
Zoals de meeste medische patronen of syndromen, waaronder EDS, is AAD een symptoomcomplex met vele onderliggende oorzaken. “Articulo” verwijst naar het gewricht, erkennend dat aandoening van elke articulaire component – huid, zenuw, spier, bot, bindweefsel, bloedvat – een gelijkaardig patroon van gewrichts-weefsel laksheid met autonome onbalans veroorzaken. Er zal broosheid van de huid zijn, verminderde botdichtheid met gevoeligheid voor fracturen, spierzwakte met pijn (myalgie), neuropathie met gevoelloosheid/tintelingen, carpale tunnel, of regionale pijnsyndromen, autonome veranderingen waaronder tachycardie, angst, niet-rustgevende slaap, onmotiliteit van de darmen, netelroos/schaafwonden, voedsel-medicatie intoleranties, allemaal in variabele mate aanwezig, zelfs binnen specifieke “types” van EDS. Ik heb geen grote verschillen gevonden tussen patiënten die passen bij bepaalde types van EDS, geïllustreerd door frequentere huidveranderingen bij type I klassieke cEDS tegenover grotere gewrichtsflexibiliteit bij type III hypermobiele hEDS. De frequenties van symptomen bij 374 patiënten geregistreerd door systematische query varieerden niet significant tussen degenen die ik diagnosticeerde als cEDS versus hEDS, en zelfs de 8 patiënten geïdentificeerd als mogelijk vasculair vEDS door het hebben van collageen type III genmutaties waren niet onderscheidend (aneurysma’s aanwezig bij één van de 8 patiënten had dezelfde lage frequentie in andere EDS categorieën). Het belangrijkste is het concept van een EDS spectrum, naar mijn mening het best weergegeven als AAD.
Herkenning van het AAD-symptomencomplex, veroorzaakt door EDS en verwante bindweefseldysplasieën, zal trieste patiëntentochten van specialist naar specialist, symptomatische diagnoses zoals duizeligheid, flauwvallen, fibromyalgie, of chronische vermoeidheid, en tragisch lijden voorkomen, terwijl er vele preventieve strategieën en behandelingen beschikbaar zijn. Bovendien verklaart de herkenning van dysautonomie de angst voor POTS en de depressie die vaak gepaard gaat met chronische pijn of activiteitsbeperking , gedragsverschillen die kunnen worden herkend als gevolgen van een medische in plaats van een psychiatrische ziekte.
Genetische testen ondersteunen het articulo-autonomische dysplasie concept
Door hun karakterisering als erfelijke aandoeningen van het bindweefsel , bleken de meer extreme vormen van EDS en verwante aandoeningen autosomaal dominante overerving te vertonen, wat betekent dat getroffen individuen één normale en één abnormale vorm van het gen (allel) hebben. Dominante overerving is zinvol omdat defect eiwit geproduceerd door het abnormale allel, analoog aan een kromme baksteen, zich zou mengen met normale bakstenen van het andere allel en een wiebelige structuur zou maken. Erkenning van het gewricht als een multi-component structuur, beperkt door de bovenliggende huid, ondersteund door inwendig bot en kraakbeen, en werkend via intrinsieke zenuwen, spieren, pezen, gewrichtsbekleding, en bloedvaten, impliceert op zijn beurt dat elk van deze elementen kan worden beïnvloed om dysplasie te produceren, een weefselverstoring die verschilt van de misvormingen veroorzaakt door andere genetische aandoeningen. De ontwikkeling van DNA klonen en sequencing zou de werking van autosomaal dominante overerving bevestigen en het vermogen van genen die enig element van de gewrichtsstructuur veranderen om het symptoompatroon van AAD te veroorzaken.
Genetische testen begonnen met een focus op een “kandidaat” gen geselecteerd vanwege de symptomen van de patiënt, geïllustreerd door het testen van het fibrilline-1 gen voor het Marfan syndroom. Het sequencen van de AGCT-letters van het gen-DNA, de “tekst” van de nucleotiden, kon substituties of “typefouten” aan het licht brengen die het aminozuur van het gecodeerde eiwit en zijn beoogde functie veranderden. Toen kwam de nieuwe technologie NextGen of rapid parallel DNA sequencing, vergelijkbaar met het in één keer lezen van alle bladzijden van een boek in plaats van van eind tot eind. Hierdoor konden gengroepen (panels) zoals die welke cardiomyopathie of bindweefseldysplasie veroorzaken of zelfs alle 23.000 genen in elke menselijke cel (ons genoom) in 2-3 maanden worden gesequeneerd. Het sequencen van de eiwitcoderende of exonische gengebieden, whole exome sequencing (WES) genoemd, kan bij veel bedrijven worden besteld en werd in mijn praktijk verkregen voor minder dan $200-300 out-of-pocket kosten bij de helft van mijn patiënten (de catalogusprijs is $9000 en het bedrag dat aan de verzekering in rekening wordt gebracht is ongeveer $20.000).
Hoewel het interpreteren van de gemiddeld 12.000 DNA-varianten gedetailleerd door WES een probleem blijft, verminderd door een groeiende epidemiologie van varianten in normale en ziektepopulaties, is de opbrengst van genveranderingen die bijdragen aan AAD-symptomen onder 620 patiënten die WES in mijn praktijk ondergaan meer dan 60%. Naast de meer dan 80 mutaties in collageengenen, waaronder die in collageen type I dat gewoonlijk geassocieerd wordt met osteogenesis imperfecta, collageen type V dat vaak geassocieerd wordt met klassieke EDS, collagenen type VI, IX en XII die oorspronkelijk beschreven werden bij myopathieën, en het gevreesde collageen type III dat geassocieerd wordt met vasculaire EDS, zijn er terugkerende mutaties in genen die huid-, zenuw-, spier-, fibrillaire en vaatcomponenten beïnvloeden die een concept van AAD versterken waarbij verandering van een gewrichtscomponent het teken- en symptoompatroon veroorzaakt dat in tabel 1 wordt geschetst. Nu EDS en verwante aandoeningen die onder het AAD concept vallen herkend worden, kan de klinische indruk ondersteund worden met objectief DNA bewijs dat counseling van patiënt en familie mogelijk maakt.
Diagnose en therapie van hypermobiliteitsstoornissen/AAD
Alertheid voor de belangrijkste symptomen van gewrichtspijn/fragiliteit en autonome (darm, hart, immuunverstoring) kan leiden tot snelle tests voor hypermobiliteit zoals getoond in figuur 1. Het duim-onderarm teken kan de hele Beighton schaal voorafgaan, waarbij 1 punt bilateraal wordt toegekend voor vingers meer dan 90 graden naar achteren gedrukt op de hand, duimen tegen onderarmen, hyperextensie van de elleboog met gestrekte armen, hyperextensie van de knieën om een achterwaartse beencurve te vormen, en 1 punt voor het vermogen om de handpalmen tegen de vloer aan te raken. Een kort onderzoek naar symptomen zoals die in tabel 1 kan het vermoeden bevestigen, eventueel geholpen door bij het lichamelijk onderzoek een aantal van de beoordelingen op te nemen die in het onderste deel van tabel 1 zijn vermeld. Merk op dat niet alle AAD patiënten lang of dun zijn, en dat een aanzienlijk deel overgewicht zal hebben als gevolg van beperkingen in activiteit door pijn en vermoeidheid. Velen zullen een elastische huid hebben, onderzocht door plooien rond de kaak of op de onderarm op te trekken, vooral wanneer dit de dunne, oppervlakkige plooien van de opperhuid zijn. Opstapeling van wit weefsel of keloïdvorming rond littekens komt vaak voor, en beoordeling van Beighton of andere hypermobiliteitsmanoeuvres die in tabel 1 staan, kan nuttig zijn. Een ingezakte houding met een voorwaartse nekcurve, scoliose en lordose kunnen worden waargenomen, samen met platvoeten of naar buiten (meer dan naar binnen) neigen bij het lopen. Bij sommige patiënten zal de spierkracht afnemen, samen met een slecht evenwicht bij tandem lopen, maar de generalist met beperkte tijd hoeft slechts op een paar van deze lichamelijke tekenen te letten; doorverwijzing zal de rest vaststellen.
Het belangrijkst zijn orthopeden die bekend zijn met de artrose en de daarmee gepaard gaande veranderingen bij EDS, zoals degeneratieve gewrichtsveranderingen, verslechterend kraakbeen, plicabanden en gebieden met dood bot. Een ervaren en conservatieve orthopeed die zich bewust is van de slechte resultaten van operaties om gewrichten “strakker” te maken of te vervangen is wenselijk. Een cardioloog kan POTS documenteren met behulp van de tilt-table test voor orthostatische veranderingen in hartslag en bloeddruk, en ook elektro- en echocardiografisch onderzoek verrichten om aortadilatatie of aneurysma’s uit te sluiten. Neurologie kan gewone migraine of dagelijkse hoofdpijn beoordelen, de Chiari vervorming die optreedt met verschuiving en herniatie van laks hersenweefsel, de neuropathie als gevolg van vernauwing binnen laxte gewrichten, en de spierveranderingen aangeduid door de symptoomomschrijving fibromyalgie en benadrukt door frequente myopathische genveranderingen die een nieuwe term van myopathische EDS in de hand werken. Allergisten die bekend zijn met mestcelactivering en gastro-enterologen die lage darmmotiliteit kunnen aantonen zijn waardevolle aanvullingen op het subspecialistische team, en niemand is waardevoller dan de huisarts voor het beheren van de vele aanbevelingen en medicatie die nodig kunnen zijn. Hoewel het vreemd kan lijken van een geneticus te komen, denk ik dat de subspecialistische evaluaties die tot therapie leiden hoger scoren dan genetische evaluatie en testen, maar de laatste kunnen nuttig zijn bij het ontkrachten van veronderstellingen over psychiatrische ziekte.
De behandeling begint met een oefenprogramma dat zowel artritis ten gevolge van hypermobiliteit van de gewrichten als de reflexieve dysautonomie ten goede kan komen. Aandacht voor gewrichtspijn (groeipijnen treden ’s nachts op) of letsel/skeletmisvorming kan preventie in gang zetten door de patiënt bewust te maken van zijn toestand (merk in tabel 1 op dat velen hun flexibiliteit niet als abnormaal herkennen). Slijtage- en scheurletsel kan worden voorkomen door kwetsbare gewrichten te verstevigen, de rug te ondersteunen met heupbanden en gewrichtsbeschermende activiteiten te kiezen, zoals zwemmen in plaats van joggen. Patiënten moeten actief blijven met matig gewichtheffen en andere redelijke activiteiten om spieren rond de gewrichten op te bouwen en cycli van inactiviteit te voorkomen die chronische vermoeidheid of fibromyalgie verergeren. Passende fysiotherapie, pijnbestrijding, oefenbegeleiding, en anekdotische medicatie zoals glucosamine/chondroïtine preparaten hebben velen geholpen, maar relaxerende middelen die de spierbescherming van gewrichten wegnemen en steroïde injecties die letsel maskeren, osteoporose/degeneratie bevorderen, en slechts tijdelijke verlichting geven, moeten worden vermeden.
De belangrijkste pijlers van de dysautonomie therapie zijn hydratatie (8-10 glazen vocht per dag), zout (afwezig labiele hypertensie), en een eiwitrijk dieet, alle gericht op het verhogen van het volume van de bloedvloeistof en het verbeteren van de cerebrale circulatie. Aanvullende voedingstherapie kan bestaan uit vitamine C (2 g per dag) vitamine D (>2000 eenheden per dag, meer dan de 400-800 in multivitaminen), vitamine B12 (1-2,5 mg per dag). Lage darmmotiliteit of gastroparese wordt bevorderd door het vermijden van vocht vóór de maaltijd, kleinere en frequentere maaltijden, probiotica, enzympreparaten en, naar verluidt van velen, minder gluten en zuivel, die vaak irriterende stoffen zijn. Oefenprogramma’s in rugligging, biofeedback, en vele medicatiemogelijkheden zijn beschikbaar via cardiologen voor POTS (b.v. bètablokkers, Midodrine, Florinef), via neurologen of pijnbestrijdingsexperts (b.v. Gabapentin, Lyrica), of via allergologen die antihistamineprotocollen kunnen aanbieden om mestcellen te onderdrukken (b.v. Zyrtec, Zantac, Cromolyn, Singulair). Voordelen van genomische testen zoals WES die verder gaan dan genen waarvan bekend is dat ze bindweefsel-dysplasie veroorzaken, zijn nieuwe bevindingen zoals de spier-gerelateerde genen waarvan nu bekend is dat ze EDS veroorzaken en documentatie van incidentele of secundaire bevindingen zoals de borst- baarmoederhalskanker genmutaties. Het vinden van genen die geassocieerd zijn met ernstiger symptomen stelt patiënten in staat risico’s voor het nageslacht te kennen en prenatale of preimplantatie genetische diagnose strategieën te kiezen die compatibel zijn met hun waarden.
Voorstelling
Eve kwam op 18-jarige leeftijd bij haar arts, duizelig, flauw, angstig en misselijk, zo uitgeput dat ze zich vrijdagavond terugtrok om het hele weekend te rusten. Ze was gezond geweest, actief in gymnastiek en cheerleaden, een dapper en slim meisje wier doorzettingsvermogen ondanks verwondingen haar coaches applaus opleverde. Toen kwam de griep waarvan ze nooit herstelde, ze werd zo misselijk dat ze niet kon eten, viel af, viel flauw in de klas, werd ’s nachts wakker met een bonkend hart, werd ’s morgens wakker zonder herstellende slaap. Eve bezocht vele dokters, zelfs de eerste hulp toen hartkloppingen en paniek haar de adem benamen. Niets aan de hand, behalve stress zeiden ze, allemaal in haar hoofd. Gekalmeerd maar niet genezen door pillen, werk mislukt door vermoeidheid en verwarring, vroeg ze zich af of ze gek aan het worden was. Niet herkend was haar zachte en kwetsbare huid, haar harde en zware menstruatie, haar dagelijkse hoofdpijn, en, het meest opvallend van alles, hypermobiliteit waarvan zij en haar familie aannamen dat het normaal was: “Ja, we zijn krom en dubbelgewrichtig, is niet iedereen dat?” zou Eve desgevraagd hebben geantwoord.
Gelukkig zocht Eve een nieuwe huisarts die veel van de in tabel I genoemde tekenen en symptomen kon documenteren. Eve had al vroeg kolieken en reflux met moeilijkheden bij het geven van borstvoeding, was zeer flexibel, door haar moeder de “botloze” baby genoemd, en toen ze ouder werd zat ze op haar knieën met haar benen gestrekt achter zich (de “W”-positie), zeggende dat het comfortabeler was. Aanvankelijk liep ze langzaam, viel vaak en leek onhandig, maar al gauw was ze actief en klom overal op, waarbij ze haar ouders deed huiveren door een voet om haar nek te leggen of met gemak de spagaat te doen. Maar toen ze naar de lagere school ging, begon ze te klagen over gewrichtspijn, waarbij haar benen ’s nachts pijn deden en haar knieën en enkels haar hinderden na het sporten. Haar kinderarts stelde haar ouders gerust dat ze gewoon “groeipijnen” had, maar ze maakten zich meer zorgen toen ze probeerde te voetballen en haar enkels bleef verzwikken, één verstuiking vereiste een laars voor 2 maanden nadat een stressfractuur was vastgesteld.
Eve moest stoppen met voetbal maar ontdekte dat ze uitblonk in turnen toen ze ouder werd, waarbij ze vloeroefeningen koos vanwege haar slechte balans op de balk en een neiging van haar schouder om uit te trekken op de parallelle staven. Ze merkte dat haar gewrichten bij het bewegen uit elkaar sprongen en deed dat vaak expres, omdat het de spanning en pijn leek te verlichten. Ze probeerde met succes cheerleaders te worden, maar ze merkte dat ze te gevoelig was voor warmte. De andere meisjes noemden haar “tomatengezicht” omdat ze oververhit raakte tijdens routines, en ze werd snel moe, vaak had ze een dag rust nodig na het sporten. Haar spijsverteringsproblemen doken weer op met regelmatige constipatie en maagpijn, vaak voelde ze zich opgeblazen en had ze laxeermiddelen nodig. In de 6e klas verzuimde ze van school vanwege haar darmproblemen en gewrichtspijn, en ze voelde zich vaak moe en duizelig. Ze voelde zich ’s morgens niet uitgerust en deed in het weekend vaak een dutje, een afwijking van haar vroegere actieve en enthousiaste levensstijl. Ze voelde zich vaak angstig, zonder dat ze dat kon herleiden tot specifieke stress op school of in haar gezin, en ze begon ’s morgens hoofdpijn te krijgen, waarvan ze dacht dat die het gevolg was van haar slechte nachtrust. Haar ouders brachten haar opnieuw naar de kinderarts en weer werd haar verteld dat er niets aan de hand was behalve de gebruikelijke schoolstress.
Toen ze 14 was had Eve een zware griepachtige ziekte en miste een maand lang school met ernstige vermoeidheid, hoofdpijn, misselijkheid, overgeven en gewichtsverlies. Ze had vreselijke duizeligheid en wat pijn op de borst, waarbij ze soms het gevoel had dat haar hart bonkte (hartkloppingen). Haar ouders vroegen een andere kinderarts om advies en die stuurde Eve naar een cardioloog, die dacht dat ze tachycardie had door stress en angst rond haar ziekte. Hij gaf Eve propranolol, een bètablokker, om haar hartslag te vertragen, maar ze werd nog vermoeider en lag nu een groot deel van de dag in bed. Haar gewrichtspijn nam toe na haar ziekte en ze bleef het grootste deel van de dag in bed liggen, voelde zich verdrietig en werd angstig als haar ouders probeerden haar mee naar buiten te nemen of een eindje te gaan rijden. Ze brachten haar terug naar de tweede kinderarts die haar doorverwees naar de psychiatrie, en er werd begonnen met Zoloft om te helpen tegen angst en depressie. Na een paar dagen kreeg Eve netelroos en moest ze stoppen met die medicatie. Ze bleef duizelig en werd door nog twee specialisten gezien, een gastro-enteroloog die dacht dat ze misschien coeliakie had en een keel-, neus- en oorarts die duizeligheid vaststelde en haar naar neurologie stuurde. Ze vertelde die arts over hoofdpijn die uit haar achterhoofd kwam, en een MRI-onderzoek van haar hoofd in rugligging was normaal.
Op grond van de waarschijnlijke diagnose AAD door haar huisarts werd een andere ronde van verwijzingen naar subspecialiteiten gestart, waaronder neurologie, waar een MRI-onderzoek van haar hoofd in rugligging de herniatie van Chiari deformatie aantoonde. Een cardioloog documenteerde een abnormale tilt-table test samen met een lage darmmotiliteit en toonde aan dat haar angst en stress-moeheid te wijten waren aan dysautonomie. Voedingsstrategieën met veel zout en eiwit, bètablokkers en antihistaminica verbeterden haar vermoeidheid en darmsymptomen en verminderden haar hoofdpijn. Er werd besloten dat de Chiari herniatie van 4-5 mm niet voldoende was om een operatie te vereisen, en haar psychiatrische behandeling werd erkend als het behandelen van secundaire symptomen van een medische stoornis. Sommige van haar subspecialisten, die sceptisch waren over haar klinische diagnose, werden overtuigd door genetische testen die een verandering aantoonden in een gen voor de alpha-1 keten van type V collageen, afgekort COL5A1, een veel voorkomende bevinding bij patiënten met EDS. Eve had de mutatie geërfd van haar moeder, die mildere AAD-symptomen had gehad en nu in staat was om ze beter te beheersen.
Over een aantal maanden waren Eve’s gezondheid en vitaliteit hersteld en kreeg ze “haar leven terug”, zoals zij en haar ouders zouden zeggen. Ze had nog steeds gewrichtspijn, angst en vermoeidheid bij gelegenheid, maar “wijze oefening” om spierbescherming rond haar gewrichten op te bouwen, voedingsbenaderingen voor dysautonomie, en biofeedback benaderingen om angst te voorkomen waren levensveranderend . Deze behandelingen waren allemaal het gevolg van de erkenning door haar huisarts, die nu in staat is om de zorg voor Eve en haar moeder te coördineren en middelen te bieden om zowel EDS als de onderliggende genveranderingen te begrijpen. De overgang naar de volgende generatie was de mogelijkheid om Eve te informeren over haar 50% risico om haar COL5A mutatie door te geven en enkele risico’s voor vroege bevalling en postpartum bloeding bij een toekomstige zwangerschap .
- Remvig L, Jensen DV, Ward RC (2007) Epidemiologie van algemene gewrichtshypermobiliteit en basis voor de voorgestelde criteria voor benigne gewrichtshypermobiliteitssyndroom: overzicht van de literatuur. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) Het Ehlers-Danlos syndroom. In: Connective Tissue and its Heritable Disorders. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomie in het gewrichtshypermobiliteitssyndroom. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lessons in pain relief–a personal postgraduate experience. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) A small molecule for a large disease. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Hoofdstuk 11: Gen- en genoomsequencing: Interpretatie van genetische variatie op het niveau van nucleotiden. In: Menselijke Chromosoom Variatie: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing–are we there yet? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Benigne joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Musculoskeletale klachten geassocieerd met gegeneraliseerde gewrichtshypermobiliteit. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Presymptomatische en preimplantatie genetische diagnostiek: Neurologie, NextGenetics, en de volgende generatie. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Behandeling van pijn en vermoeidheid bij het gewrichtshypermobiliteitssyndroom (ook bekend als het Ehlers- Danlos syndroom, hypermobiliteitstype): principes en voorstel voor een multidisciplinaire aanpak. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Multidisciplinaire behandeling van invaliditeit bij het Ehlers-Danlos syndroom hypermobiliteit type/ hypermobiliteitssyndroom: een pilotstudie met een combinatie van fysieke en cognitieve gedragstherapie bij 12 vrouwen. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exoomanalyse van bindweefseldysplasie: dood en wedergeboorte van de klinische genetica? Am J Med Genet A 164A: 1209-1212.