Słowa kluczowe
hypermobility, connective tissue dysplasia, ehlers-danlos syndrome, beighton scale, dysautonomia, postural orthostatic tachycardia syndrome, zespół jelita drażliwego, zaburzenie aktywacji komórek tucznych, dysplazja artykulacyjno-autonomiczna
Wprowadzenie
Bardzo słabo rozpoznane i tragicznie źle leczone jest 20% kobiet i 10% mężczyzn, których tkanki są bardziej elastyczne i kruche niż przeciętnie. Niewiele możliwości zapobiegania i leczenia przewyższa rozpoznanie hipermobilności, którą łatwo sprawdzić, prosząc pacjenta o oparcie kciuka o przedramię (ryc. 1). Ten znak ostrzegawczy może wskazać drogę do bardziej szczegółowej oceny, która leży w gestii lekarza ogólnego, a jego szeroka wiedza kliniczna jest dobrze dopasowana do wielosystemowych problemów, które mogą wynikać z rozluźnienia tkanki łącznej. W tym miejscu dokonuję przeglądu natury zaburzeń hipermobilności, wykazuję, że wiedza ogólna, a nie subspecjalistyczna, może prowadzić do terapii zmieniających życie tych pacjentów i pokazuję, w jaki sposób badania genetyczne wprowadzają opisy objawowe, takie jak fibromialgia czy zespół przewlekłego zmęczenia, w ostre światło medycyny precyzyjnej.
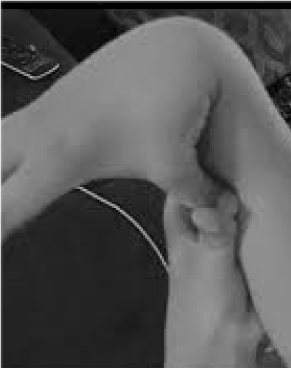
Rysunek 1. Hipermobilność.
Kilka barier musi zostać pokonanych, zanim pacjenci z hipermobilnością będą mogli czerpać korzyści z medycyny ogólnej. Pierwsza z nich wiąże się z pionierskim, ale krótkowzrocznym podejściem dermatologów Ehlersa i Danlosa, którzy odcisnęli piętno na niewiarygodnym i egzotycznym rozciągnięciu skóry jako mylącym symbolu zaburzeń wiotkości tkanki łącznej. Drugą barierą jest rosnąca przepaść między medycyną praktyczną i genetyczną, z których pierwsza odpowiednio koncentruje się na powszechnych chorobach, podczas gdy druga do niedawna wykorzystywała specjalistyczną technologię do opisywania rzadkich. Niejasne eponimy, zniechęcające podtypy i terminy techniczne są jak małe prowincje nieciekawego obcego kraju, których żaden globalny podróżnik nie poświęci czasu na mapowanie. Przeciwdziałanie tym przeszkodom polega na praktycznym podejściu do zaburzeń hipermobilności, które koncentruje się na rozpoznaniu na podstawie wywiadu i badania fizykalnego, pozostawiając techniczne aspekty eponimów, podtypów i badań genetycznych subspecjalistom, z którymi kontaktuje się po istotnym pierwszym etapie wstępnej diagnozy. Po rozpoznaniu i skierowaniu elastyczny pacjent pozostaje zależny od swojego lekarza pierwszego kontaktu, ponieważ tylko on może zintegrować zalecenia i wyniki badań z ogólnym domem medycznym.
Terminologia
Zwiększony zakres ruchu w wielu miejscach artykulacji (stawów) występuje w dużej liczbie chorób, począwszy od zaburzeń chromosomowych, takich jak zespół Downa, do zaburzeń takich jak zespół Ehlersa-Danlosa (EDS), znanych z hipermobilności. McKusick spopularyzował tę kategorię zaburzeń znanych z luźności tkanek, grupując je jako dziedziczne zaburzenia tkanki łącznej, w tym zespół Marfana, na którym skupił się jako kardiolog. W 1977 r. dr Peter Beighton, znany ze swojej skali mierzącej giętkość, uporządkował dotychczasową literaturę, postulując 6-7 typów EDS, obecnie uproszczonych w najnowszych wytycznych jako EDS klasyczne, naczyniowe i hipermobilne. Jest teraz jasne, że te typy mają znaczne nakładanie się objawów między sobą i powiązanych zaburzeń, takich jak osteogeneza niedoskonała (z licznymi złamaniami), zespół Marfana (duże rozszerzenie/rozprężenie naczyń), zespół Sticklera (zmiany oczu i stawów) i wiele innych.
Raczej niż zajmować się poszczególnymi chorobami, uważam, że bardziej przydatne jest skupienie się na dwóch głównych procesach chorobowych z dysplazji tkanki łącznej, zilustrowanych przez wiele objawów wymienionych w Tabeli 1. Pierwszym z nich jest zużywający się ból stawów (choroba zwyrodnieniowa stawów), który różni się od obrzękniętych, czerwonych stawów zapalenia stawów (autoimmunologicznego lub zapalnego) spotykanego w reumatoidalnym zapaleniu stawów, toczniu, zespole Sjogrena lub zesztywniającym zapaleniu stawów kręgosłupa. Po drugie, jest to zaburzenie równowagi autonomicznego układu nerwowego (dysautonomia) spowodowane przez elastyczne naczynia krwionośne w zaburzeniach hipermobilności. Rozszerzające się naczynia powodują gromadzenie się krwi w miednicy i nogach, z obniżeniem ciśnienia krwi i zawrotami głowy/omdleniami w pozycji stojącej (niedociśnienie ortostatyczne). Z czasem zmniejszony przepływ krwi do głowy aktywuje współczulne ramię autonomicznego układu nerwowego (ramię adrenaliny „uciekaj lub walcz”), co prowadzi do kilku problemów: 1) zespół posturalnej tachykardii ortostatycznej (POTS) z tachykardią, niepokojem, chronicznym zmęczeniem i „mgłą mózgową” (okresy zmniejszonej koncentracji i pamięci), 2) zespół jelita drażliwego z powodu zahamowania motoryki jelit (supresja przywspółczulna) z zaparciami na przemian z biegunkami, wzdęcia, refluks, bóle brzucha, dysfagia, mdłości i 3) zaburzenia aktywacji komórek tucznych z przejściowymi wysypkami, pokrzywką/reaktywną skórą, astmą/dusznością i nietolerancją pokarmów/leków. Opracowałem szerszy termin, który ułatwia rozpoznawanie wszelkich zaburzeń hipermobilności, uznając dwa główne następstwa wiotkości stawów/tkanek: dysplazję stawowo-autonomiczną lub zespół zmęczenia stawów.
Tabela 1. Typowe wyniki badań i ich częstość w zespołach hipermobilności. *Od 320 kobiet i 54 mężczyzn w wieku powyżej 12 lat ocenianych za pomocą szczegółowych kwestionariuszy w 2017 roku; TMJ, staw skroniowo-żuchwowy; POTS, zespół posturalnej tachykardii ortostatycznej. Lista objawów zaadaptowana częściowo z Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
HISTORIA (procenty)*
W niemowlęctwie: Kolka (28), trudności z karmieniem (19,4); niezdarność, liczne upadki (29,1)
Dzieci/nastolatki: Powolny przyrost masy ciała (12); świadomy elastyczności (55,6); popisywał się sztuczkami stawowymi (53,6)
wczesne bóle stawów (42,1); aktywności nastolatków ograniczone przez ból-kontuzje (55,4); wczesne okulary (46,8)
ortodoncja (61,4%) choroby dziąseł (11,3); słabe szkliwo/wiele ubytków (31,6)
Problemy ze stawami: Popping (81.6); podluksacje (62); TMJ popping, catching, pain (51.2)
ból w co najmniej 2 stawach (80.3); operacje stawów (33); wiele zwichnięć (26); złamania >2 (31.5)
przepuklina dysku kręgosłupa (29.3)
Problemy z układem kostnym: Skolioza (31); pectus (2,2); chód – wdech/wydech (9,3); płaskostopie (15,5)
Skóra: Łatwe powstawanie siniaków (61); rozciągliwa skóra (15); aksamitna (18); nietypowe blizny (36,3)
rozstępy przed ciążą (73); powolne gojenie (32,4)
Genitourinary (females): Menorrhagia (61,2); endometrioza (11,3); torbiele jajników (55)
zespół policystycznych jajników (8,5); problemy z pęcherzem moczowym (46,1); przepuklina pachwinowa/śródpiersiowa (11,5)
Neuromięśniowe: Migreny (51,7); codzienne bóle głowy (62,3); potrzebny lek na ból głowy (45)
Zniekształcenie Chiari (10,6); operacja neurochirurgiczna (2,9); drętwienie/ mrowienie (48,7); słaba równowaga (48,6)
neuropatyczne bóle strzelające (37,3); bóle/skurcze mięśni (51,7); osłabienie mięśni (32,4)
Żywienie/jelita: Zaparcia-biegunka (67,1); wzdęcia-refluks-ból brzucha (57,3)
problemy z pęcherzykiem żółciowym (12,9); utrudnione połykanie (23); częste nudności (44,9)
Serce: Wypadanie płatka zastawki mitralnej (8,7); tętniak (0,9); arytmia inna niż tachykardia (6,8)
POTY: Zawroty głowy w pozycji stojącej (69,8); omdlenia (34,5); przewlekłe zmęczenie (81,8); trudności ze snem (43,5)
Mgła mózgu (słaba pamięć lub skupienie uwagi w niektórych momentach-70,8); wrażliwość na ciepło/zimno (77,5); nieprawidłowa potliwość (25); tachykardia (72,8); napady lęku/paniki (61,3); ochota na sól (51,2)
Komórka immunologiczna/mastocytoma: Przemijające wysypki (33,4%); pokrzywka/reaktywna skóra (45,9%); astma/duszność (39,3); nietolerancje pokarmów/leków (61,5%)
Lab/Imaging: Niska gęstość kości (2,1); niska witamina D (28,7); niska witamina B12 (2,7); niedoczynność tarczycy (11,5); niska ferrytyna (1,7); niski poziom żelaza (6,7); podwyższone przeciwciała przeciwjądrowe (7.1)
Średnia liczba wyników badań na 80: kobiety >12 (35,4 wyników) mężczyźni >12 (21,5 wyników)
fizyczne (w procentach)
Budowa: Wysoki – wzrost >90 percentyl (55,2); ciężki-BMI >28 (21,3); szczupły-BMI <19 (14,2)
Habitus marfanoidalny (44); arachnodaktylia (46,4); objaw Walkera-Murdocha obecny (40,2)
Twarz: Długa (25,7); wąska dolna część twarzy (5,4); niebieskoszara twardówka (1,6); wysokie podniebienie (53,5)
Skóra: Miękka (81,4); półprzezroczysta (23,8); elastyczno-lift >1-calowy fałd wokół szczęki na śródręczu (51,3)
Fałdy składają się z warstwy zewnętrznej (naskórkowej) (35.4); nietypowe blizny (42,1%)
Punktacja Beightona: 0-3 punkty (8,4); 4-6 punktów (32,6); 7-8 punktów (38,8); 9 z 9 punktów (20,2)
Inne manewry: Złączenie rąk nad barkiem-za plecami (68)
znak modlitwy w górę za plecami (71,3); przeniesienie ręki wokół pleców, aby dotknąć pępka (19,5)
Szkieletowe: Kifoza szyjna (41,2); skolioza (22,3); lordoza (32,8); płaskostopie (39,6)
chód – wdech lub wydech (28,3)
Neurologiczne: Osłabienie mięśni (11,2); zmniejszona masa mięśniowa (8,8)
słaba równowaga przez chód tandemowy (33,4)
Średnia liczba ustaleń na 40: kobiety >12 (17,9 ustaleń) mężczyźni >12 (15.7 wyników)
Dysplazja artykulo-autonomiczna (AAD)
Termin taki jak „dysplazja artykulo-autonomiczna” lub AAD może ujednolicić podejście do zaburzeń wiotkości tkanki łącznej/hipermobilności i przeciwdziałać trzem problemom, które komplikują ich rozpoznanie: 1) wielu lekarzy i genetyków postrzega dysplazje tkanki łącznej jako choroby rzadkie i ekstremalne, podczas gdy w rzeczywistości dotykają one około 1% populacji 2) wytyczne diagnostyczne EDS, choć upraszczają główne typy EDS jako klasyczne, naczyniowe i hipermobilności, nadal wprowadzają w błąd, odnosząc się do wielu pomniejszych typów, które tworzą nakładające się spektrum objawów, oraz 3) prawie obowiązkowa dysautonomia, która towarzyszy EDS i pokrewnym zaburzeniom (Tabela 1) jest pomijana jako kryterium diagnostyczne, analogicznie do opisywania słonia bez wymieniania jego trąby. AAD oznacza nieunikniony fizjologiczny łącznik pomiędzy luźną tkanką i dysautonomią w ten sam sposób, w jaki zespoły metaboliczne lub hepatorenalne opisują jak otyłość prowadzi do choroby wątroby/cukrzycy lub niewydolność wątroby powoduje chorobę nerek.
Jak większość medycznych wzorców lub zespołów, włączając EDS, AAD jest kompleksem objawów z wieloma podstawowymi przyczynami. „Articulo” odnosi się do stawu, uznając, że dolegliwości jakiegokolwiek składnika stawowego – skóry, nerwu, mięśnia, kości, tkanki łącznej, naczynia krwionośnego – powodują podobny wzór luźności tkanki stawowej z zaburzeniem równowagi autonomicznej. Będzie kruchość skóry, zmniejszona gęstość kości z podatnością na złamania, osłabienie mięśni z bólem (mialgia), neuropatia z drętwieniem/mrowieniem, cieśń nadgarstka, lub regionalne zespoły bólowe, zmiany autonomiczne w tym tachykardia, niepokój, sen nierestoracyjny, nieruchliwość jelit, pokrzywka/ wysypka, nietolerancja pokarmów i leków, wszystkie obecne w różnym stopniu nawet w określonych „typach” EDS. Nie stwierdziłem dużych różnic między pacjentami z poszczególnymi typami EDS, czego przykładem są częstsze zmiany skórne w typie I klasycznym cEDS i większa elastyczność stawów w typie III hipermobilnym hEDS. Częstości objawów u 374 pacjentów zarejestrowanych przez systematyczną kwerendę nie różniły się znacząco pomiędzy tymi, których zdiagnozowałem jako cEDS versus hEDS, a nawet 8 pacjentów zidentyfikowanych jako możliwe naczyniowe vEDS poprzez posiadanie mutacji genu kolagenu typu III nie wyróżniało się niczym szczególnym (tętniaki obecne u jednego z 8 pacjentów miały tak samo niską częstotliwość w innych kategoriach EDS). Najważniejsza jest koncepcja spektrum EDS, najlepiej reprezentowana moim zdaniem jako AAD.
Rozpoznanie kompleksu objawów AAD, spowodowanego przez EDS i pokrewne dysplazje tkanki łącznej, zapobiegnie smutnym podróżom pacjentów od specjalisty do specjalisty, objawowym diagnozom, takim jak zawroty głowy, omdlenia, fibromialgia lub przewlekłe zmęczenie, oraz tragicznemu cierpieniu, gdy dostępnych jest wiele strategii zapobiegawczych i metod leczenia. Ponadto, rozpoznanie dysautonomii wyjaśnia niepokój związany z POTS i depresję, która często towarzyszy przewlekłemu bólowi lub ograniczeniu aktywności, różnice w zachowaniu, które mogą być uznane za konsekwencje choroby medycznej, a nie psychiatrycznej.
Badania genetyczne wspierają koncepcję dysplazji stawowo – autonomicznej
Poparte ich charakterystyką jako dziedzicznych zaburzeń tkanki łącznej, bardziej ekstremalne formy EDS i zaburzeń pokrewnych okazały się wykazywać dziedziczenie autosomalne dominujące, co oznacza, że osoby dotknięte chorobą mają jedną normalną i jedną nieprawidłową formę genu (allel). Dziedziczenie dominujące ma sens, ponieważ wadliwe białko produkowane przez nieprawidłowy allel, analogicznie do wypaczonej cegły, miesza się z normalnymi cegłami pochodzącymi z drugiego allelu i tworzy chybotliwą strukturę. Uznanie stawu lub połączenia stawowego za strukturę wieloskładnikową, ograniczaną przez pokrywającą go skórę, podtrzymywaną przez wewnętrzną tkankę kostną i chrzęstną oraz działającą poprzez wewnętrzne nerwy, mięśnie, ścięgna, wyściółkę stawu i naczynia krwionośne, oznacza z kolei, że każdy z tych elementów może ulec wpływowi, który doprowadzi do powstania dysplazji, zaburzenia tkanki różniącego się od wad rozwojowych spowodowanych innymi zaburzeniami genetycznymi. Rozwój klonowania i sekwencjonowania DNA potwierdziłby działanie dziedziczenia autosomalnego dominującego i zdolność genów zmieniających dowolny element struktury stawu do wywoływania objawów AAD.
Badania genetyczne rozpoczęły się od skupienia uwagi na genie „kandydującym”, wybranym ze względu na objawy występujące u pacjenta, co ilustruje badanie genu fibryliny-1 w zespole Marfana. Sekwencjonowanie liter AGCT DNA genu, „tekstu” nukleotydów, mogło ujawnić substytucje lub „literówki”, które zmieniły aminokwas kodowanego białka i jego zamierzoną funkcję. Wtedy pojawiła się nowa technologia zwana NextGen lub szybkie równoległe sekwencjonowanie DNA, analogiczne do czytania wszystkich stron książki na raz, a nie od końca do końca. Umożliwiło to sekwencjonowanie grup genów (paneli), takich jak te powodujące kardiomiopatię lub dysplazję tkanki łącznej, a nawet wszystkich 23 000 genów w każdej ludzkiej komórce (naszym genomie) w ciągu 2-3 miesięcy. Sekwencjonowanie regionów kodujących białka lub egzonicznych genów, zwane sekwencjonowaniem całego eksomu (WES), można zamówić w wielu firmach i w mojej praktyce zostało uzyskane za mniej niż 200-300 USD kosztów zewnętrznych u połowy moich pacjentów (cena katalogowa wynosi 9000 USD, a kwota rozliczana z ubezpieczeniem wynosi około 20 000 USD).
Ale chociaż interpretacja średnio 12,000 wariantów DNA wyszczególnionych przez WES pozostaje problemem, zmniejszonym przez rosnącą epidemiologię wariantów w populacjach normalnych i chorobowych, wydajność zmian genów przyczyniających się do objawów AAD wśród 620 pacjentów poddanych WES w mojej praktyce wynosi ponad 60%. Oprócz ponad 80 mutacji genów kolagenu, w tym tych w kolagenie typu I zwykle związanym z osteogenesis imperfecta, kolagenie typu V często związanym z klasycznym EDS, kolagenach typu VI, IX i XII początkowo opisanych z miopatiami, oraz kolagenie typu III związanym z EDS naczyniowym, są powtarzające się mutacje w genach wpływających na skórę, nerwy, mięśnie, elementy fibrylarne i naczynia, które wzmacniają koncepcję AAD, gdzie zmiana dowolnego składnika stawu wytwarza znak i wzór objawów przedstawiony w Tabeli 1. Obecnie rozpoznanie EDS i pokrewnych zaburzeń objętych koncepcją AAD może wzmocnić wrażenie kliniczne obiektywnymi dowodami DNA, które umożliwiają poradnictwo dla pacjenta i jego rodziny.
Diagnostyka i terapia zaburzeń hipermobilności/AAD
Uwaga na główne objawy bólu stawów/zwiotczenia i zaburzeń autonomicznych (jelit, serca, układu odpornościowego) może prowadzić do szybkich testów w kierunku hipermobilności, jak przedstawiono na rycinie 1. Znak „kciuk do przedramienia” może być wstępem do całej skali Beightona, dając jeden punkt obustronnie za palce wciśnięte do tyłu poza 90 stopni na dłoni, dotykanie kciukami przedramion, zginanie łokcia przy wyprostowanych rękach, zginanie kolan do tyłu i 1 punkt za zdolność dotykania dłońmi podłogi. Krótki przegląd objawów, takich jak te w Tabeli 1, może potwierdzić podejrzenie, wspomagany być może przez włączenie do badania fizykalnego niektórych z ocen wymienionych w dolnej części Tabeli 1. Należy pamiętać, że nie wszyscy pacjenci z AAD są wysocy lub szczupli, a znaczna część będzie miała nadwagę z powodu ograniczeń aktywności wynikających z bólu i zmęczenia. Wielu z nich będzie miało elastyczną skórę, co można sprawdzić podciągając fałdy wokół żuchwy lub na przedramieniu, szczególnie jeśli są to cienkie, powierzchowne fałdy naskórka. Nagromadzenie białej tkanki lub tworzenie się keloidów wokół blizn jest powszechne, a ocena Beightona lub innych manewrów hipermobilności wymienionych w tabeli 1 może być pomocna. Można zaobserwować pochyloną postawę z wygięciem szyi do przodu, skoliozą i lordozą, a także płaskostopie lub wysklepienie stóp (bardziej niż do wewnątrz) podczas chodzenia. Siła mięśni będzie obniżona u niektórych pacjentów wraz ze słabą równowagą podczas chodzenia w tandemie, ale lekarz ogólny z ograniczonym czasem musi zwrócić uwagę tylko na kilka z tych objawów fizycznych; skierowanie ustali resztę.
Najważniejsi będą ortopedzi zaznajomieni z chorobą zwyrodnieniową stawów i towarzyszącymi jej zmianami w EDS, takimi jak zmiany zwyrodnieniowe stawów, pogarszająca się chrząstka, pasma plica i regiony martwej kości. Doświadczony i konserwatywny ortopeda, który rozpoznaje słabe wyniki operacji w celu „dokręcenia” lub zastąpienia stawów jest pożądane. Kardiolog może udokumentować POTS za pomocą testu tilt-table dla ortostatycznych zmian w częstości akcji serca i ciśnienia krwi, wykonując również elektro- i echokardiograficzne badania w celu wykluczenia rozszerzenia aorty lub tętniaków. Neurologia może ocenić powszechne migreny lub codzienne bóle głowy, deformację Chiari, która występuje z poślizgiem i przepukliną luźnej tkanki mózgowej, neuropatię spowodowaną zwężeniem w obrębie luźnych stawów oraz zmiany w mięśniach oznaczane przez opis objawów fibromialgii i podkreślane przez częste miopatyczne zmiany w genach, co sprzyja rozwojowi nowego terminu miopatycznego EDS. Alergolodzy zaznajomieni z aktywacją komórek tucznych i gastroenterolodzy, którzy mogą udokumentować niską motorykę jelit są cennymi dodatkami do zespołu podspecjalistycznego, a nikt nie jest bardziej wartościowy niż lekarz pierwszego kontaktu w zarządzaniu wieloma zaleceniami i lekami, które mogą być wymagane. Chociaż może się to wydawać dziwne, że pochodzi od genetyka, uważam, że oceny podspecjalistyczne, które prowadzą do terapii, oceniają wyżej niż oceny i badania genetyczne, ale te ostatnie mogą być pomocne w odrzuceniu założeń dotyczących choroby psychicznej.
Leczenie rozpoczyna się od programu ćwiczeń, który może przynieść korzyść zarówno zapaleniu stawów spowodowanemu hipermobilnością stawów, jak i odruchowej dysautonomii. Zwracanie uwagi na bóle stawów (bóle wzrostowe występują w nocy) lub urazy/deformacje szkieletowe może zapoczątkować profilaktykę poprzez uświadomienie pacjentowi jego stanu (w Tabeli 1 zaznaczono, że wiele osób nie rozpoznaje swojej elastyczności jako nieprawidłowej). Urazom związanym ze zużyciem można zapobiegać poprzez usztywnienie wrażliwych stawów, założenie opasek na plecy oraz wybór aktywności chroniących stawy, takich jak pływanie czy jogging. Pacjenci powinni pozostać aktywni poprzez umiarkowane podnoszenie ciężarów i inne rozsądne działania mające na celu budowanie mięśni wokół stawów, zapobieganie cyklom braku aktywności, które nasilają przewlekłe zmęczenie lub fibromialgię. Odpowiednia fizykoterapia, leczenie bólu, wskazówki dotyczące ćwiczeń i niepotwierdzone leki, takie jak preparaty glukozaminy/chondroityny pomogły wielu osobom, ale należy unikać środków zwiotczających, które odbierają mięśniom ochronę stawów i zastrzyków sterydowych, które maskują uraz, promują osteoporozę/degenerację i dają tylko tymczasową ulgę.
Podstawą terapii dysautonomii jest nawodnienie (8-10 szklanek płynu dziennie), sól (nieobecne labilne nadciśnienie) i dieta wysokobiałkowa, wszystkie mające na celu zwiększenie objętości płynu we krwi i poprawę krążenia mózgowego. Dodatkowe leczenie żywieniowe może obejmować witaminę C (2 g dziennie) witaminę D (>2000 jednostek dziennie, więcej niż 400-800 w multiwitaminach), witaminę B12 (1-2,5 mg dziennie). Niska motoryka jelit lub gastropareza jest wspomagana przez unikanie płynów przed posiłkami, mniejsze i częstsze posiłki, probiotyki, preparaty enzymatyczne i, według raportu wielu, obniżenie glutenu i nabiału, które są częstymi czynnikami drażniącymi. Supine programy ćwiczeń, biofeedback, i wiele opcji leków są dostępne przez kardiologów dla POTS (np., beta-blokery, Midodrine, Florinef), przez neurologów lub ekspertów zarządzania bólem (np., Gabapentin, Lyrica), lub przez alergologów, którzy mogą zapewnić protokoły antyhistaminowe do tłumienia komórek tucznych (np., Zyrtec, Zantac, Cromolyn, Singulair). Zaletami badań genomowych, takich jak WES, które wykraczają poza geny znane jako przyczyny dysplazji tkanki łącznej, są nowe odkrycia, takie jak geny związane z mięśniami, o których obecnie wiadomo, że powodują EDS, oraz dokumentacja przypadkowych lub wtórnych odkryć, takich jak mutacje genów raka piersi i jajnika. Znalezienie genów związanych z poważniejszymi objawami pozwala pacjentom poznać ryzyko dla potomstwa i wybrać strategie prenatalnej lub preimplantacyjnej diagnostyki genetycznej zgodne z ich wartościami.
Prezentacja przypadku
Eve przyszła do swojego lekarza w wieku 18 lat, z zawrotami głowy, omdleniami, niepokojem i chorobą żołądka, tak wyczerpana, że przeszła na emeryturę w piątek wieczorem, aby odpocząć przez cały weekend. Była zdrowa, aktywna w gimnastyce i cheerleaderkach, odważna i bystra dziewczyna, której wytrwałość mimo kontuzji przyniosła jej aplauz trenerów. Potem grypa, z której nigdy się nie wyleczyła, mdłości tak silne, że nie mogła jeść, utrata wagi, omdlenia w klasie, budzenie się w nocy z walącym sercem, budzenie się rano bez regenerującego snu. Ewa była u wielu lekarzy, nawet na ostrym dyżurze, kiedy kołatanie serca i panika odbierały jej oddech. Mówili, że nic jej nie dolega, że to tylko stres, że wszystko jest w jej głowie. Uspokojona, ale nie wyleczona tabletkami, zawodząca w pracy z powodu zmęczenia i dezorientacji, zastanawiała się, czy nie oszalała. Nie rozpoznano jej miękkiej i delikatnej skóry, twardych i obfitych miesiączek, codziennych bólów głowy i, co najbardziej uderzające, hipermobilności, którą ona i jej rodzina uznali za normalną: „Tak, jesteśmy zgięci i dwustawowi, czyż nie wszyscy?”. Ewa odpowiedziałaby, gdyby ją zapytano.
Na szczęście Ewa poszukała nowego lekarza pierwszego kontaktu, który był w stanie udokumentować wiele oznak i objawów wymienionych w Tabeli I. Ewa miała wczesną kolkę i refluks z trudnościami w karmieniu piersią, była bardzo giętka, nazywana przez matkę dzieckiem „bez kości”, a gdy była starsza, siadała na kolanach z nogami wyciągniętymi za siebie (pozycja „W”), twierdząc, że tak jest wygodniej. Początkowo wolno chodziła, często się przewracała i sprawiała wrażenie niezdarnej, ale wkrótce była aktywna i wszędzie się wspinała, przyprawiając rodziców o ból głowy, gdy zakładała nogę na szyję lub z łatwością robiła pompki. Ale gdy weszła do szkoły podstawowej, zaczęła skarżyć się na bóle stawów, nogi bolały ją w nocy, a kolana i kostki dokuczały jej po wysiłku. Jej pediatra uspokoił jej rodziców, że po prostu miała „bóle wzrostowe”, ale stali się bardziej zaniepokojeni, gdy próbowała grać w piłkę nożną i ciągle przewracała kostki, jedno zwichnięcie wymagające buta przez 2 miesiące po tym, jak zauważono złamanie.
Eve musiała przestać grać w piłkę nożną, ale okazało się, że świetnie radzi sobie z gimnastyką, gdy dorastała, wybierając ćwiczenia na podłodze z powodu słabej równowagi na belce i tendencji do wyciągania ramienia na równoległych barach. Zauważyła, że jej stawy wyskakują przy ruchu i często wyskakiwała je celowo, ponieważ wydawało się, że łagodzi to napięcie i ból. Próbowała swoich sił w cheerleadingu, odnosząc sukcesy w manewrach, ale okazało się, że jest zbyt wrażliwa na ciepło. Inne dziewczyny nazywały ją „pomidorową twarzą”, ponieważ przegrzewała się podczas ćwiczeń i łatwo się męczyła, często potrzebując dnia odpoczynku po ćwiczeniach. Jej problemy trawienne powróciły z częstymi zaparciami i bólem brzucha, często czuła się wzdęta i potrzebowała środków przeczyszczających. W szóstej klasie opuszczała lekcje z powodu problemów z jelitami i bólów stawów, często czuła się zmęczona i miała zawroty głowy. Rano nie czuła się wypoczęta i często ucinała sobie drzemki w weekendy, co stanowiło odejście od jej wcześniejszego aktywnego i entuzjastycznego stylu życia. Często czuła się niespokojna, nie mogąc powiązać tego z żadnym konkretnym stresem szkolnym lub rodzinnym, a rano zaczęła mieć bóle głowy, o których myślała, że są spowodowane słabym snem. Jej rodzice ponownie zabrali ją do pediatry i ponownie powiedziano im, że nie ma nic złego, ale to zwykły stres szkolny.
Kiedy miała 14 lat Ewa miała ciężką chorobę grypopodobną i opuściła szkołę na miesiąc z silnym zmęczeniem, bólami głowy, nudnościami, wymiotami i utratą wagi. Miała straszne zawroty głowy i bóle w klatce piersiowej, czasami czuła, że jej serce wali (miała palpitacje). Rodzice poradzili się innego pediatry, a ten wysłał Ewę do kardiologa, który uznał, że tachykardia jest wynikiem stresu i niepokoju związanego z jej chorobą. Podał Ewie propranolol, beta-bloker, aby zwolnić akcję serca, ale ona stała się jeszcze bardziej zmęczona i przez większość dnia była przykuta do łóżka. Bóle stawów nasiliły się po chorobie i Ewa przeleżała w łóżku większość dnia, czując smutek i niepokój, kiedy rodzice próbowali ją wyprowadzić na dwór lub zabrać na przejażdżkę. Zanieśli ją z powrotem do drugiego pediatry, który skierował ją do psychiatry i rozpoczęto podawanie Zoloftu, który miał pomóc w leczeniu lęku i depresji. Po kilku dniach Ewa dostała wysypki z pokrzywką i musiała odstawić ten lek. W dalszym ciągu miała zawroty głowy i trafiła do dwóch kolejnych specjalistów: gastroenterologa, który uznał, że może mieć celiakię i laryngologa, który zdiagnozował zawroty głowy i wysłał ją na neurologię. Powiedziała temu lekarzowi o bólach głowy emanujących z tyłu głowy i wykonano badanie MRI głowy w pozycji leżącej, które było prawidłowe.
Docenienie prawdopodobnej diagnozy AAD przez jej lekarza pierwszego kontaktu zapoczątkowało inną rundę skierowań do podspecjalistów, w tym neurologa, gdzie badanie MRI głowy w pozycji stojącej wykazało przepuklinę deformacji Chiari. Kardiolog udokumentował nieprawidłowy test tilt-table wraz z niską motoryką jelit i udowodnił, że jej niepokój i zmęczenie spowodowane stresem wynikały z dysautonomii. Strategie żywieniowe polegające na nawadnianiu, spożywaniu soli i dużej ilości białka oraz terapia beta-blokerami i lekami przeciwhistaminowymi poprawiły jej zmęczenie i objawy jelitowe oraz zmniejszyły bóle głowy. Zdecydowano, że przepuklina Chiariego o wielkości 4-5 mm nie jest wystarczająca, aby wymagać operacji, a jej leczenie psychiatryczne uznano za leczenie objawów wtórnych do zaburzeń medycznych. Niektórzy z jej subspecjalistów, sceptycznie nastawieni do diagnozy klinicznej, zostali przekonani przez badania genetyczne, które udokumentowały zmianę w genie dla łańcucha alfa-1 kolagenu typu V, w skrócie COL5A1, co jest częstym wynikiem u pacjentów z EDS. Ewa odziedziczyła tę mutację po swojej matce, która miała łagodniejsze objawy AAD i teraz była w stanie lepiej sobie z nimi radzić.
W ciągu kilku miesięcy zdrowie i witalność Ewy zostały przywrócone, a ona sama „odzyskała swoje życie”, jak powiedzieliby jej rodzice. Nadal miała bóle stawów, niepokój i zmęczenie od czasu do czasu, ale „mądre ćwiczenia”, aby zbudować ochronę mięśni wokół jej stawów, metody żywieniowe dla dysautonomii i metody biofeedback, aby zapobiec niepokojowi, zmieniły jej życie. Wszystkie te zabiegi były wynikiem uznania przez jej lekarza rodzinnego, który teraz jest w stanie koordynować opiekę nad Ewą i jej matką oraz zapewnić środki do zrozumienia zarówno EDS jak i zmian genowych leżących u jego podstaw. Przejściem do następnego pokolenia była możliwość poinformowania Ewy o jej 50% ryzyku przekazania mutacji COL5A i pewnych zagrożeń dla wczesnego porodu i krwawienia poporodowego w każdej przyszłej ciąży.
- Remvig L, Jensen DV, Ward RC (2007) Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: review of the literature. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. In: Tkanka łączna i jej dziedziczne zaburzenia. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomia w zespole hipermobilności stawów. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lessons in pain relief–a personal postgraduate experience. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) A small molecule for a large disease. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Rozdział 11: Sekwencjonowanie genów i genomów: Interpretacja zmienności genetycznej na poziomie nukleotydów. In: Human Chromosome Variation: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing–are we there yet? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Benign joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Musculoskeletal complaints associated with generalized joint hypermobility. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Presymptomatyczna i preimplantacyjna diagnostyka genetyczna: Neurologia, NextGenetics i następne pokolenie. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Management of pain and fatigue in the joint hypermobility syndrome (a.k.a. Ehlers- Danlos syndrome, hypermobility type): principles and proposal for a multidisciplinary approach. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Multidisciplinary treatment of disability in Ehlers-Danlos syndrome hypermobility type/zespół hipermobilności: a pilot study using a combination of physical and cognitivebehavioral therapy on 12 women. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exome analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am J Med Genet A 164A: 1209-1212.