Historie
La începutul anilor 1900, cercetătorii au descoperit că includerea unor cantități mari de albuș de ou crud în alimentația șobolanilor a produs simptome de toxicitate. În 1926, Boas s-a referit la aceste simptome de toxicitate ca fiind sindromul de rănire a albușului de ou. Principalele constatări au inclus dermatită severă, pierderea părului și lipsa de coordonare musculară. Boas a observat, de asemenea, că drojdia, ficatul și alte câteva produse alimentare conțineau o substanță care proteja șobolanii de sindromul leziunilor cauzate de albușul de ou. O căutare a acestui factor de protecție a dus la descoperirea, în 1936, a biotinei.
Bazele biochimice ale sindromului de rănire a albușului de ou au fost rapid elucidate atunci când s-a constatat că albușul de ou crud conține glicoproteina avidină, care are o afinitate remarcabilă pentru biotină. Legătura biotină-avidină este în esență ireversibilă; ca urmare, biotina nu este eliberată din alimente, iar complexul biotină-avidină se pierde în fecale. Pasul final în rezolvarea misterului sindromului leziunilor cauzate de albușul de ou a fost demonstrarea faptului că sindromul poate fi prevenit prin încălzirea albușurilor de ou, un proces care denaturează avidina și îi distruge afinitatea pentru biotină.
Structura
Biotina este o moleculă biciclică compusă dintr-un inel ureido fuzionat cu un inel tetrahidrotiofen.
Inelul ureido este implicat în legătura de mare afinitate a biotinei cu avidina, o glicoproteină care se găsește în albușul de ou. Un substituent acid valeric este atașat la unul dintre cei 2 atomi de carbon ai inelului tetrahidrotiofenic. Prin această grupare carboxil, biotina este legată covalent de grupa β-amino a lizinei în 5 carboxilaze care joacă roluri critice în metabolismul intermediar.
FUNCȚIILE BIOTINULUI
În plus față de rolul bine cunoscut al biotinei ca și cofactor în reacțiile de carboxilare, studii recente au arătat că biotina joacă roluri importante în reglarea expresiei genice și a funcției imunitare.
Reacțiile de carboxilare dependente de biotină
Biotina funcționează ca o coenzimă în reacțiile de carboxilare care implică metabolismul lipidelor, glucozei și aminoacizilor. Există 5 carboxilaze dependente de biotină, fiecare dintre ele existând ca apoformă inactivă. Enzima holocarboxilaza sintetază (HCLS ) catalizează adăugarea de biotină (biotinilare) la apoforma inactivă, ceea ce duce la formarea carboxilazei active. În toate cele 5 carboxilaze, biotina funcționează ca o coenzimă sau grup protetic care servește ca transportor pentru CO2 într-o reacție în mai multe etape.
Cele cinci carboxilaze dependente de biotină și funcțiile lor sunt descrise pe scurt mai jos:
Piruvat carboxilaza (PC) catalizează formarea oxaloacetatului din piruvat, o etapă importantă în ciclul TCA, gluconeogeneză și lipogeneză; lipsa acestei funcții poate duce la hipoglicemie, cetoză și acidoză lactică.
Propionil-CoA carboxilaza (PCC) catalizează conversia propionil CoA în metilmalonil CoA, care la rândul său se izomerizează în succinil CoA și intră în ciclul TCA (Kreb). PCC este importantă în metabolismul acizilor grași cu lanț impar și al aminoacizilor izoleucină, valină, metionină și treonină. Lipsa acestei funcții enzimatice poate duce la acidemia propionică. Nivelurile de PCC în limfocite este un indicator sensibil al statusului de biotină.
3-Metilcrotonoil-CoA carboxilaza (MCC) este implicată în catabolismul aminoacidului cu lanț ramificat, leucina. Lipsa biotinei poate duce la devierea produșilor catabolismului leucinei într-o cale catabolică alternativă care duce la producerea de acid 3-hidoxiizovaleric care este apoi excretat în urină.
Acetil-CoA carboxilaza I (ACC I) catalizează conversia acetil CoA în malonil CoA, în citosol, o etapă importantă în sinteza lipidelor.
Acetil-CoA carboxilaza II (ACC II), catalizează o reacție identică în mitocondrii; malonil CoA rezultată joacă un rol reglator în oxidarea acizilor grași.
ACC I este o enzimă citosolică; restul carboxilazelor se găsesc în mitocondrii.
Manifestările clinice ale deficitului de biotină pot apărea, de asemenea, ca urmare a tulburărilor genetice care cauzează o deficiență a enzimei holocarboxilaza sintetază sau deficiențe ale enzimelor carboxilazei individuale.
Expresia genică
Studii au arătat că biotinilarea histonelor poate juca un rol în expresia genică. În histonele umane au fost identificate multiple situsuri care se leagă de biotină. Biotina poate afecta, de asemenea, expresia genelor prin alte mecanisme. În celulele umane sunt cunoscute câteva mii de gene dependente de biotină. Printre genele influențate de biotină se numără cele care codifică pentru enzimele implicate în metabolismul glucozei (de exemplu, glucokinaza), citokine precum interleukina-2 și receptorul de insulină.
Funcția imunitară
Studiile sugerează un rol al biotinei în producția de anticorpi, în funcția macrofagică, în diferențierea limfocitelor T și B, precum și în funcția normală a celulelor natural killer. Infecțiile recurente, în special cele fungice, sunt frecvente la pacienții cu deficit de biotină.
Rolul biotinei în afecțiunile neurologice care răspund la doze mari de biotină
Boala ganglionilor bazali care răspunde la biotină (BTBGD) este o afecțiune neurologică rară care se poate prezenta cu convulsii și encefalopatie care progresează până la comă și deces. Doza mare de biotină ( 5-10 mg/kg/zi) a fost utilizată pentru a trata cu succes această afecțiune, dar mecanismul de acțiune este necunoscut.
Recent, s-a constatat că tratamentul cu doze mari de biotină (100-300 mg/zi) îmbunătățește simptomele la un subgrup de pacienți cu scleroză multiplă. Se crede că ameliorarea simptomelor neurologice poate implica o îmbunătățire a producției de mielină secundară efectului biotinei în doze mari asupra sintezei acizilor grași cu lanț lung.
Rolul biotinei la persoanele cu afecțiuni ale părului, pielii și unghiilor
Suplimentele cu biotină sunt utilizate pe scară largă de către cei care speră să obțină un păr, o piele și unghii mai sănătoase. Cu toate acestea, există dovezi limitate cu privire la eficacitatea biotinei pentru această utilizare. S-a constatat că biotina în doze mari este utilă în cazul a două afecțiuni rare: sindromul familial al părului incomode și sindromul unghiilor fragile. Într-un studiu efectuat pe 541 de femei care prezentau căderea părului, s-a constatat că nivelurile serice de biotină erau scăzute la 38% dintre pacienți. Autorul a concluzionat că etiologia căderii părului este multifactorială și că suplimentele de biotină pot fi luate în considerare în cazul în care deficitul de biotină a fost demonstrat și alte cauze au fost excluse.
Surse de biotină
Biotina este prezentă într-o mare varietate de alimente (carne, produse lactate, legume, semințe și nuci) și este, de asemenea, produsă de bacteriile intestinale. În plus, o proporție substanțială de persoane poate consuma suplimente alimentare care conțin biotină.
Fiziologia biotinei
Biotina ingerată este prezentă în forme libere și legate de proteine. Formele legate de proteine sunt digerate de proteazele și peptidazele gastrointestinale pentru a forma biotină și biotin-oligopeptide. Biotina liberă este eliberată din biocitină și biotină-oligopeptide prin acțiunea biotinidazei intestinale. Biotina liberă este apoi absorbită în intestinul subțire prin intermediul unui mecanism mediat de transport dependent de Na+, care transportă, de asemenea, alte două substanțe nutritive, acidul pantotenic și lipoatul și, prin urmare, este cunoscut sub numele de transportor de multivitamine dependent de sodiu (SMVT). Gena SMVT umană este localizată pe cromozomul 2p23. Activitatea SMVT este reglată de nivelurile de biotină, fiind reglată în sus în cazul deficitului de biotină și în jos în cazul suplimentării excesive cu biotină. Biotina sintetizată pe cale bacteriană este prezentă sub formă nelegată și este absorbită în intestinul gros printr-un mecanism similar mediat de transport. Producția zilnică combinată de biotină în urină și în scaun depășește aportul alimentar de biotină, ceea ce sugerează rolul important jucat de flora intestinală ca sursă de biotină.
După ce este absorbită, biotina devine disponibilă pentru diverse procese de biotinilare. Biotina se leagă de fiecare dintre cele 5 apocarboxilaze pentru a forma holocarboxilaza corespunzătoare (a se vedea imaginea de mai jos) prin acțiunea enzimei holocarboxilaza sintetază.
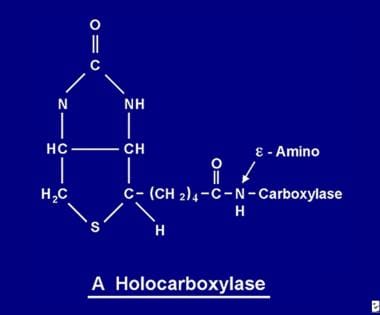 Molecula de biotină este legată de proteină printr-o legătură peptidică la o grupare e-amino a unei apocarboxilaze pentru a forma o holocarboxilază.
Molecula de biotină este legată de proteină printr-o legătură peptidică la o grupare e-amino a unei apocarboxilaze pentru a forma o holocarboxilază. Reciclarea biotinei
După ce enzima holocarboxilază a efectuat mai multe carboxilații, aceasta este captată de lizozomii celulari. În lizozomi, diverse enzime proteolitice degradează holocarboxilaza pentru a forma biotină, care, la rândul ei, este hidrolizată de enzima biotinidază pentru a forma biotină și lizină. Biotina liberă este apoi disponibilă pentru a fi inserată într-o apocarboxilază pentru a forma o nouă moleculă de holocarboxilază. Acest proces de reciclare nu este 100% eficient. Ca urmare, cantități mici de biotină liberă (și o parte din biocitină) scapă din ciclu și se pierd în fecale și urină. Din acest motiv, biotina trebuie să fie furnizată în intestin, pentru a reface biotina pierdută din organism. Etapele implicate în reciclarea biotinei – intrarea acesteia în intestin, absorbția sa, încorporarea sa în holocarboxilaze, care la rândul lor sunt descompuse pentru a elibera biotina liberă – constituie ciclul biotinei și este descris în imaginea de mai jos.
Enzima biotinidază este esențială pentru reciclarea biotinei și, prin urmare, persoanele cu deficit de biotinidază vor prezenta semne și simptome de deficit de biotină.
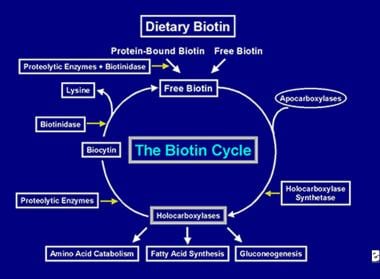 Reprezentare a fluxului de biotină în ciclul biotinei.
Reprezentare a fluxului de biotină în ciclul biotinei. CAUZELE DEFICIENȚEI DE BIOTINA
După cum s-a menționat anterior, biotina este disponibilă pe scară largă în alimente, este, de asemenea, produsă de flora intestinală și este reciclată pe scară largă în organism cu ajutorul enzimei biotinidaza; prin urmare, deficiența de biotină la indivizii sănătoși care au o dietă normală este rară. Condițiile care pot duce la deficit de biotină sunt descrise mai jos:
Consumul excesiv de albuș de ou crud: Avidina din albușul de ou crud are o afinitate mare pentru biotină, făcând-o indisponibilă pentru absorbție. Căldura distruge avidina, astfel încât cei care consumă ouă fierte nu sunt expuși riscului de deficiență de biotină. Albușul de ou crud duce la deficiență de biotină doar atunci când este consumat în cantități excesive (poate o duzină sau mai multe pe zi).
Nutriție parenterală totală fără suplimentarea cu biotină: Au fost raportate mai multe cazuri de deficiență de biotină la pacienții care au primit terapie prelungită de nutriție parenterală totală (TPN) fără adaos de biotină. Prin urmare, toți pacienții care primesc TPN trebuie să primească, de asemenea, biotină în doza zilnică recomandată, în special dacă se preconizează că terapia TPN va dura mai mult de 1 săptămână. Toate farmaciile din spitale includ în prezent biotina în preparatele TPN.
Utilizarea de formule pentru sugari cu biotină inadecvată: A fost raportată o deficiență de biotină la sugarii care primesc formule hipoalergenice.
Terapie cu anticonvulsivante cronice: Utilizarea prelungită a anticonvulsivantelor, fenobarbital, fenitoină, primidonă și carbamazepină, a fost legată de deficitul de biotină. Printre mecanismele posibile se numără inhibarea absorbției biotinei prin mucoasa intestinală, catabolismul accelerat al biotinei și afectarea reabsorbției renale a biotinei. Prin urmare, a fost sugerat un supliment de biotină pentru pacienții care sunt tratați cu anticonvulsivante care au fost legate de deficitul de biotină.
Terapie antibiotică orală prelungită: Utilizarea prelungită a antibioticelor orale a fost asociată cu deficiența de biotină. Se presupune că inhibarea florei intestinale care produce biotină este la baza deficienței de biotină. Un alt mecanism posibil ar putea fi creșterea excesivă, indusă de antibiotice, a bacteriilor care consumă biotină.
Fumat și alcoolism cronic: Studiile au arătat că fumatul poate accelera catabolismul biotinei, în special la femei. Alcoolismul cronic poate cauza malabsorbția intestinală a biotinei.
Sindromul intestinului scurt și boala inflamatorie intestinală: Persoanele cu sindromul intestinului scurt și boala inflamatorie intestinală sunt, de asemenea, la risc de deficit de biotină ca urmare a malabsorbției intestinale a biotinei.
Deficiență marginală de biotină în timpul sarcinii și alăptării: Studii recente au arătat niveluri scăzute de biotină la o proporție semnificativă de femei însărcinate și care alăptează. Există temeri că deficiența marginală de biotină în timpul sarcinii ar putea fi teratogenă și unii experți au recomandat un aport mai mare de biotină de către femeile însărcinate.
Certe erori înnăscute ale metabolismului biotinei pot duce, de asemenea, la manifestarea deficitului de biotină.
Deficiența de biotinidază (BTD) se moștenește în mod autosomal recesiv și apare la o frecvență de aproximativ 1 la 60.000 de nașteri vii; se estimează că 1 din 120 de indivizi sunt heterozigoți pentru această afecțiune. La indivizii homozigoți pentru această afecțiune, nivelurile de biotinidază sunt < 30% normale, ceea ce duce la deficit de biotină din cauza eliberării insuficiente de biotină liberă din cauza scăderii reciclării biotinei. Simptomele BTD se dezvoltă de obicei între 1 săptămână și 1 an de vârstă. Severitatea deficienței enzimatice poate varia. Cei cu deficit profund de biotinidază au niveluri de BTD mai mici de 10% normal, în timp ce cei cu deficit parțial de biotinidază au niveluri enzimatice între 10-30% normal. În SUA și în multe alte țări, screeningul nou-născuților include teste pentru deficitul de BTD. Aproximativ 150 de mutații în gena BTD au fost raportate ca fiind cauza deficitului de biotinidază. Gena BTD este localizată pe cromozomul 3p25. A fost dezvoltat un model de șoarece de deficit de biotinidază pentru a studia diverse aspecte ale acestei afecțiuni.
Deficiența deolocarboxilază sintetază (HCLS) este, de asemenea, o tulburare autozomal recesivă și poate fi diagnosticată prenatal. După cum s-a discutat anterior, enzima HCLS este necesară pentru biotinilarea enzimelor apocarboxilaza în formele active de holocarboxilază; prin urmare, deficitul duce la deficit de carboxilază multiplă. Sugarii cu această afecțiune se prezintă în primele luni de viață cu acidoză, hiperamoniemie, hipotonie, convulsii și întârziere în dezvoltare. Mutațiile în gena HCLS determină deficitul de HCLS.
Pentru că atât deficitul de biotinidază, cât și cel de HCLS duc la scăderea nivelurilor carboxilazelor dependente de biotină, cele două afecțiuni au fost clasificate și ca deficit de carboxilază multiplă. Deficitul profund de biotinidază era cunoscut anterior ca deficit de carboxilaza multiplă cu debut precoce, deficitul parțial de biotinidază ca deficit de carboxilaza multiplă cu debut tardiv sau juvenil și deficitul de HCLS ca deficit de carboxilaza multiplă neonatal sau cu debut precoce.
Rar, pot apărea, de asemenea, deficiențe izolate ale fiecăreia dintre cele cinci carboxilaze individuale dependente de biotină.
Au fost descrise, de asemenea, deficiențe de biotină datorate unui defect în transportul biotinei.
Indiferent de etiologia deficitului de biotină, manifestările clinice sunt similare. Cu toate acestea, vârsta de debut, rata de dezvoltare a simptomelor și succesiunea în care apar simptomele pot fi foarte diferite. Toate mecanismele responsabile pentru dezvoltarea manifestărilor nu au fost stabilite.
.