Geschichte
In den frühen 1900er Jahren stellten Forscher fest, dass die Aufnahme großer Mengen rohen Eiweißes in die Ernährung von Ratten zu Symptomen der Toxizität führte. Im Jahr 1926 bezeichnete Boas diese Vergiftungserscheinungen als „egg-white injury syndrome“. Zu den wichtigsten Befunden gehörten schwere Dermatitis, Haarausfall und mangelnde muskuläre Koordination. Boas stellte auch fest, dass Hefe, Leber und verschiedene andere Nahrungsmittel eine Substanz enthielten, die Ratten vor dem Eiweißverletzungssyndrom schützte. Die Suche nach diesem Schutzfaktor führte 1936 zur Entdeckung von Biotin.
Die biochemische Grundlage des Eiweißverletzungssyndroms war schnell geklärt, als man feststellte, dass rohes Eiweiß das Glykoprotein Avidin enthält, das eine bemerkenswerte Affinität zu Biotin hat. Die Biotin-Avidin-Bindung ist im Wesentlichen irreversibel; daher wird Biotin nicht aus der Nahrung freigesetzt, und der Biotin-Avidin-Komplex geht mit den Fäkalien verloren. Der letzte Schritt zur Lösung des Rätsels des Eiweißverletzungssyndroms war der Nachweis, dass das Syndrom durch Erhitzen des Eiweißes verhindert werden kann, ein Prozess, der Avidin denaturiert und seine Affinität für Biotin zerstört.
Struktur
Biotin ist ein bicyclisches Molekül, das aus einem Ureidoring besteht, der mit einem Tetrahydrothiophenring verschmolzen ist.
Der Ureidoring ist an der hohen Affinität von Biotin zu Avidin, einem Glykoprotein im Eiklar, beteiligt. Ein Valeriansäure-Substituent ist an eines der beiden Kohlenstoffatome des Tetrahydrothiophenrings gebunden. Durch diese Carboxylgruppe ist Biotin kovalent an die β-Aminogruppe von Lysin in 5 Carboxylasen gebunden, die eine entscheidende Rolle im Intermediärstoffwechsel spielen.
FUNKTIONEN VON BIOTIN
Neben der bekannten Rolle von Biotin als Cofaktor bei Carboxylierungsreaktionen haben neuere Studien gezeigt, dass Biotin eine wichtige Rolle bei der Regulierung der Genexpression und der Immunfunktion spielt.
Biotin-abhängige Carboxylierungsreaktionen
Biotin fungiert als Coenzym bei Carboxylierungsreaktionen im Lipid-, Glucose- und Aminosäurestoffwechsel. Es gibt 5 biotinabhängige Carboxylasen, von denen jede in einer inaktiven Apoform vorliegt. Das Enzym Holocarboxylase-Synthetase (HCLS ) katalysiert die Anlagerung von Biotin (Biotinylierung) an die inaktive Apoform, was zur Bildung der aktiven Carboxylase führt. In allen 5 Carboxylasen fungiert Biotin als Coenzym oder prosthetische Gruppe, die als Träger für CO2 in einer mehrstufigen Reaktion dient.
Die fünf biotinabhängigen Carboxylasen und ihre Funktionen werden im Folgenden kurz beschrieben:
Pyruvatcarboxylase (PC) katalysiert die Bildung von Oxalacetat aus Pyruvat, ein wichtiger Schritt für den TCA-Zyklus, die Gluconeogenese und die Lipogenese; ein Fehlen dieser Funktion kann zu Hypoglykämie, Ketose und Laktatazidose führen.
Propionyl-CoA-Carboxylase (PCC) katalysiert die Umwandlung von Propionyl-CoA in Methylmalonyl-CoA, das wiederum zu Succinyl-CoA isomerisiert und in den TCA-Zyklus (Kreb-Zyklus) gelangt. PCC ist wichtig für den Stoffwechsel der ungeradkettigen Fettsäuren und der Aminosäuren Isoleucin, Valin, Methionin und Threonin. Ein Mangel an dieser Enzymfunktion kann zu Propionsäureanämie führen. Der PCC-Spiegel in Lymphozyten ist ein empfindlicher Indikator für den Biotinstatus.
3-Methylcrotonoyl-CoA-Carboxylase (MCC) ist am Abbau der verzweigtkettigen Aminosäure Leucin beteiligt. Ein Biotinmangel kann dazu führen, dass Leucinabbauprodukte in einen alternativen Abbaupfad umgelenkt werden, was zur Bildung von 3-Hydoxyisovaleriansäure führt, die dann mit dem Urin ausgeschieden wird.
Acetyl-CoA-Carboxylase I (ACC I) katalysiert die Umwandlung von Acetyl-CoA in Malonyl-CoA im Cytosol, ein für die Lipidsynthese wichtiger Schritt.
Acetyl-CoA-Carboxylase II (ACC II) katalysiert eine identische Reaktion in den Mitochondrien; das entstehende Malonyl-CoA spielt eine regulierende Rolle bei der Fettsäureoxidation.
ACC I ist ein zytosolisches Enzym; die übrigen Carboxylasen befinden sich in den Mitochondrien.
Klinische Manifestationen von Biotinmangel können auch als Folge genetischer Störungen auftreten, die einen Mangel des Enzyms Holocarboxylase-Synthetase oder Mängel der einzelnen Carboxylase-Enzyme verursachen.
Genexpression
Studien haben gezeigt, dass die Biotinylierung von Histonen eine Rolle bei der Genexpression spielen kann. In menschlichen Histonen wurden mehrere Stellen identifiziert, die an Biotin binden. Biotin kann die Genexpression auch durch andere Mechanismen beeinflussen. In menschlichen Zellen sind einige tausend biotinabhängige Gene bekannt. Zu den Genen, die von Biotin beeinflusst werden, gehören solche, die für Enzyme des Glukosestoffwechsels (z. B. Glucokinase), Zytokine wie Interleukin-2 und den Insulinrezeptor kodieren.
Immunfunktion
Studien deuten auf eine Rolle von Biotin bei der Antikörperproduktion, der Makrophagenfunktion, der Differenzierung von T- und B-Lymphozyten sowie der normalen Funktion von natürlichen Killerzellen hin. Wiederkehrende Infektionen, insbesondere Pilzinfektionen, treten bei Patienten mit Biotinmangel häufig auf.
Rolle von Biotin bei neurologischen Erkrankungen, die auf eine hohe Biotin-Dosis ansprechen
Die auf Biotin reagierende Basalganglienerkrankung (BTBGD) ist eine seltene neurologische Erkrankung, die mit Krampfanfällen und Enzephalopathie bis hin zum Koma und Tod einhergehen kann. Hochdosiertes Biotin (5-10 mg/kg/Tag) wurde zur erfolgreichen Behandlung dieser Erkrankung eingesetzt, aber der Wirkungsmechanismus ist unbekannt.
In jüngster Zeit wurde festgestellt, dass eine hochdosierte Biotinbehandlung (100-300 mg/Tag) die Symptome bei einer Untergruppe von Patienten mit Multipler Sklerose verbessert. Es wird vermutet, dass die Verbesserung der neurologischen Symptome mit einer verbesserten Myelinproduktion einhergeht, die auf die Wirkung von hochdosiertem Biotin auf die Synthese langkettiger Fettsäuren zurückzuführen ist.
Rolle von Biotin bei Personen mit Haar-, Haut- und Nagelstörungen
Biotin-Nahrungsergänzungen werden häufig von Personen eingenommen, die sich gesündere Haare, Haut und Nägel wünschen. Die Wirksamkeit von Biotin für diesen Zweck ist jedoch nur begrenzt belegt. Biotin in hohen Dosen hat sich bei zwei seltenen Erkrankungen als hilfreich erwiesen: beim familiären Syndrom der nicht biegsamen Haare und beim Syndrom der brüchigen Nägel. In einer Studie mit 541 Frauen, die unter Haarausfall litten, wurde bei 38 % der Patientinnen ein niedriger Biotinspiegel im Serum festgestellt. Die Autorin kam zu dem Schluss, dass die Ätiologie des Haarausfalls multifaktoriell ist und Biotinzusätze in Betracht gezogen werden können, wenn ein Biotinmangel nachgewiesen wurde und andere Ursachen ausgeschlossen wurden.
Quellen von Biotin
Biotin ist in einer Vielzahl von Lebensmitteln (Fleisch, Milchprodukte, Gemüse, Samen und Nüsse) enthalten und wird auch von Darmbakterien produziert. Darüber hinaus nimmt ein erheblicher Teil der Menschen biotinhaltige Nahrungsergänzungsmittel zu sich.
Biotinphysiologie
Das aufgenommene Biotin liegt in freier und proteingebundener Form vor. Die proteingebundenen Formen werden durch gastrointestinale Proteasen und Peptidasen verdaut und bilden Biocytin und Biotin-Oligopeptide. Freies Biotin wird aus Biocytin und Biotin-Oligopeptiden durch die Wirkung der intestinalen Biotinidase freigesetzt. Freies Biotin wird dann im Dünndarm über einen Na+-abhängigen, Carrier-vermittelten Mechanismus absorbiert, der auch zwei andere Nährstoffe, Pantothensäure und Lipoat, transportiert und daher als Natrium-abhängiger Multivitamintransporter (SMVT) bekannt ist. Das menschliche SMVT-Gen befindet sich auf dem Chromosom 2p23. Die SMVT-Aktivität wird durch den Biotinspiegel reguliert, wobei sie bei Biotinmangel hochreguliert und bei Biotinübersupplementierung herunterreguliert wird. Bakteriell synthetisiertes Biotin liegt in ungebundener Form vor und wird im Dickdarm durch einen ähnlichen Carrier-vermittelten Mechanismus absorbiert. Die kombinierte tägliche Ausscheidung von Biotin im Urin und im Stuhl übersteigt die Aufnahme von Biotin mit der Nahrung, was auf die wichtige Rolle der Darmflora als Biotinquelle hinweist.
Nach der Resorption wird Biotin für verschiedene Biotinylierungsprozesse verfügbar. Biotin bindet sich an jede der 5 Apocarboxylasen, um durch die Wirkung des Enzyms Holocarboxylase-Synthetase die entsprechende Holocarboxylase (siehe Abbildung unten) zu bilden.
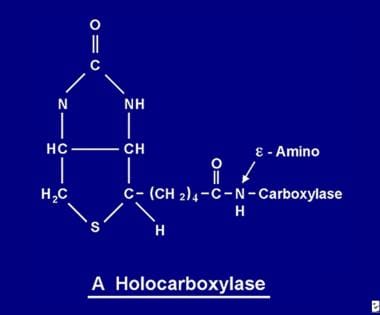 Das Biotinmolekül wird durch eine Peptidbindung an eine e-Aminogruppe einer Apocarboxylase an das Protein gebunden, um eine Holocarboxylase zu bilden.
Das Biotinmolekül wird durch eine Peptidbindung an eine e-Aminogruppe einer Apocarboxylase an das Protein gebunden, um eine Holocarboxylase zu bilden. Wiederverwertung von Biotin
Nachdem das Holocarboxylase-Enzym mehrere Carboxylierungen durchgeführt hat, wird es von zellulären Lysosomen aufgefangen. In den Lysosomen bauen verschiedene proteolytische Enzyme die Holocarboxylase ab, um Biocytin zu bilden, das wiederum durch das Enzym Biotinidase zu Biotin und Lysin hydrolysiert wird. Das freie Biotin steht dann für den Einbau in eine Apocarboxylase zur Bildung eines neuen Holocarboxylase-Moleküls zur Verfügung. Dieser Recyclingprozess ist nicht zu 100 % effizient. Infolgedessen entgehen kleine Mengen an freiem Biotin (und etwas Biocytin) dem Kreislauf und gehen mit den Fäkalien und dem Urin verloren. Aus diesem Grund muss dem Darm Biotin zugeführt werden, um das dem Körper entzogene Biotin wieder aufzufüllen. Die am Biotin-Recycling beteiligten Schritte – sein Eintritt in den Darm, seine Absorption, sein Einbau in Holocarboxylasen, die ihrerseits abgebaut werden, um freies Biotin freizusetzen – bilden den Biotin-Zyklus und sind in der folgenden Abbildung dargestellt.
Das Enzym Biotinidase ist für das Biotin-Recycling unerlässlich, weshalb Personen mit Biotinidase-Mangel Anzeichen und Symptome eines Biotinmangels aufweisen.
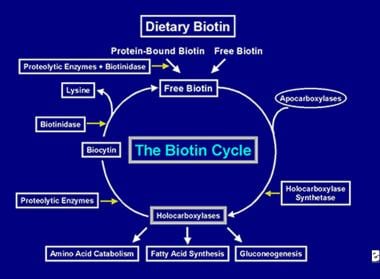 Darstellung des Biotinflusses im Biotin-Zyklus.
Darstellung des Biotinflusses im Biotin-Zyklus. Ursachen von Biotinmangel
Wie bereits erwähnt, ist Biotin in Lebensmitteln reichlich vorhanden, wird auch von der Darmflora produziert und im Körper mit Hilfe des Enzyms Biotinidase weitgehend recycelt; daher ist Biotinmangel bei gesunden Menschen, die sich normal ernähren, selten. Bedingungen, die zu Biotinmangel führen können, werden im Folgenden beschrieben:
Übermäßiger Verzehr von rohem Eiweiß: Das Avidin in rohem Eiweiß hat eine hohe Affinität für Biotin, wodurch es nicht aufgenommen werden kann. Hitze zerstört das Avidin, so dass Menschen, die gekochte Eier essen, nicht von einem Biotinmangel bedroht sind. Rohes Eiweiß führt nur dann zu Biotinmangel, wenn es in übermäßigen Mengen gegessen wird (vielleicht ein Dutzend oder mehr pro Tag).
Totale parenterale Ernährung ohne Biotinsupplementierung: Es wurde über mehrere Fälle von Biotinmangel bei Patienten berichtet, die über einen längeren Zeitraum eine totale parenterale Ernährung (TPN) ohne Biotinzusatz erhielten. Daher müssen alle Patienten, die TPN erhalten, auch Biotin in der empfohlenen Tagesdosis erhalten, insbesondere wenn die TPN-Therapie voraussichtlich länger als eine Woche dauert. Alle Krankenhausapotheken enthalten derzeit Biotin in TPN-Zubereitungen.
Verwendung von Säuglingsnahrung mit unzureichendem Biotin: Es wurde über Biotinmangel bei Säuglingen berichtet, die hypoallergene Nahrung erhielten.
Chronische Antikonvulsivum-Therapie: Die längerfristige Einnahme der Antikonvulsiva Phenobarbital, Phenytoin, Primidon und Carbamazepin wurde mit Biotinmangel in Verbindung gebracht. Zu den möglichen Mechanismen gehören die Hemmung der Biotinaufnahme über die Darmschleimhaut, ein beschleunigter Biotinkatabolismus und eine gestörte Rückresorption von Biotin über die Nieren. Daher wird eine zusätzliche Biotinzufuhr für Patienten empfohlen, die mit Antikonvulsiva behandelt werden, die mit Biotinmangel in Verbindung gebracht wurden.
Längere orale Antibiotikatherapie: Die längere Einnahme von oralen Antibiotika wurde mit Biotinmangel in Verbindung gebracht. Als Ursache für den Biotinmangel wird eine Hemmung der biotinbildenden Darmflora vermutet. Ein weiterer möglicher Mechanismus könnte eine durch Antibiotika verursachte Überwucherung von Bakterien sein, die Biotin verbrauchen.
Rauchen und chronischer Alkoholismus: Studien haben gezeigt, dass Rauchen den Biotinkatabolismus beschleunigen kann, insbesondere bei Frauen. Chronischer Alkoholismus kann zu einer intestinalen Malabsorption von Biotin führen.
Kurzdarmsyndrom und entzündliche Darmerkrankungen: Bei Personen mit Kurzdarmsyndrom und entzündlichen Darmerkrankungen besteht ebenfalls das Risiko eines Biotinmangels als Folge einer intestinalen Malabsorption von Biotin.
Marginaler Biotinmangel während Schwangerschaft und Stillzeit: Jüngste Studien haben verminderte Biotinwerte bei einem signifikanten Anteil von schwangeren und stillenden Frauen gezeigt. Es gibt Bedenken, dass ein marginaler Biotinmangel während der Schwangerschaft teratogen sein könnte, und einige Experten haben eine höhere Biotinzufuhr für Schwangere empfohlen.
Bestimmte angeborene Fehler des Biotin-Stoffwechsels können ebenfalls zur Manifestation eines Biotinmangels führen.
Biotinidase-Mangel (BTD) wird autosomal rezessiv vererbt und tritt mit einer Häufigkeit von etwa 1 von 60.000 Lebendgeburten auf; schätzungsweise 1 von 120 Personen ist heterozygot für die Erkrankung. Bei Personen, die homozygot für die Störung sind, ist der Biotinidase-Spiegel < 30 % normal, was zu einem Biotinmangel führt, der auf eine unzureichende Freisetzung von freiem Biotin aufgrund eines verminderten Biotin-Recyclings zurückzuführen ist. Die Symptome der BTD entwickeln sich typischerweise im Alter von 1 Woche bis 1 Jahr. Der Schweregrad des Enzymmangels kann variieren. Bei Patienten mit schwerem Biotinidase-Mangel liegt der BTD-Spiegel unter 10 % des Normalwerts, während bei Patienten mit partiellem Biotinidase-Mangel der Enzymspiegel zwischen 10 und 30 % des Normalwerts liegt. In den USA und vielen anderen Ländern umfasst das Neugeborenenscreening Tests auf BTD-Mangel. Etwa 150 Mutationen im BTD-Gen sind bekannt, die einen Biotinidase-Mangel verursachen. Das BTD-Gen befindet sich auf Chromosom 3p25. Es wurde ein Mausmodell des Biotinidase-Mangels entwickelt, um verschiedene Aspekte der Störung zu untersuchen.
Holocarboxylase-Synthetase (HCLS)-Mangel ist ebenfalls eine autosomal rezessive Störung und kann pränatal diagnostiziert werden. Wie bereits erwähnt, ist das Enzym HCLS für die Biotinylierung der Apocarboxylase-Enzyme zu den aktiven Holocarboxylase-Formen erforderlich; ein Mangel führt daher zu einem Mangel an multiplen Carboxylasen. Säuglinge mit dieser Störung zeigen in den ersten Lebensmonaten Azidose, Hyperammonämie, Hypotonie, Krampfanfälle und Entwicklungsverzögerungen. Mutationen im HCLS-Gen verursachen einen HCLS-Mangel.
Da sowohl der Biotinidase- als auch der HCLS-Mangel zu verminderten Spiegeln der biotinabhängigen Carboxylasen führt, werden die beiden Erkrankungen auch als multipler Carboxylasemangel bezeichnet. Ein ausgeprägter Biotinidase-Mangel wurde früher als früh einsetzender multipler Carboxylase-Mangel bezeichnet, ein partieller Biotinidase-Mangel als spät einsetzender oder juveniler multipler Carboxylase-Mangel und ein HCLS-Mangel als neonataler oder früh einsetzender multipler Carboxylase-Mangel.
Selten können auch isolierte Defizite jeder der fünf einzelnen biotinabhängigen Carboxylasen auftreten.
Biotinmangel aufgrund eines Defekts im Biotintransport ist ebenfalls beschrieben worden.
Unabhängig von der Ätiologie des Biotinmangels sind die klinischen Manifestationen ähnlich. Das Alter des Ausbruchs, die Geschwindigkeit der Symptomentwicklung und die Reihenfolge, in der die Symptome auftreten, können jedoch sehr unterschiedlich sein. Nicht alle Mechanismen, die für die Entwicklung der Manifestationen verantwortlich sind, sind bekannt.