Palabras clave
hipermovilidad, displasia del tejido conectivo, síndrome de ehlers-danlos, escala de beighton, disautonomía, síndrome de taquicardia ortostática postural, síndrome del intestino irritable, trastorno de activación de mastocitos, displasia articular-autonómica
Introducción
El 20% de las mujeres y el 10% de los hombres cuyos tejidos son más flexibles y frágiles que la media están muy poco reconocidos y trágicamente mal tratados. Pocas oportunidades de prevención y tratamiento superan la de reconocer la hipermovilidad, que se puede detectar fácilmente pidiendo al paciente que empuje el pulgar contra el antebrazo (figura 1). Este signo revelador puede indicar el camino hacia una evaluación más detallada que está dentro del ámbito del médico general, ya que sus amplios conocimientos clínicos coinciden con los problemas multisistémicos que pueden surgir del tejido conectivo laxo. Aquí reviso la naturaleza de los trastornos de hipermovilidad, demuestro que el conocimiento general, más que el de la subespecialidad, puede conducir a terapias que cambien la vida de estos pacientes, y muestro cómo las pruebas genéticas traen descripciones sintomáticas como la fibromialgia o el síndrome de fatiga crónica a la luz del discernimiento de la medicina de precisión.
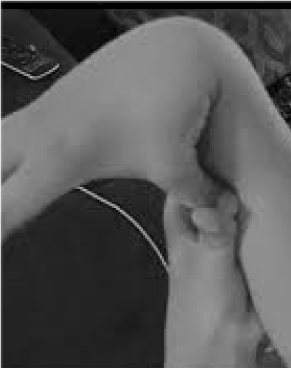
Figura 1. Hipermovilidad.
Hay que superar varias barreras antes de que los pacientes con hipermovilidad puedan obtener los beneficios de la medicina general. La primera deriva implica el enfoque pionero pero miope de los dermatólogos Ehlers y Danlos imprimiendo un increíble y exótico estiramiento de la piel como símbolo engañoso de los trastornos de laxitud del tejido conectivo. Una segunda barrera es la creciente división entre la medicina práctica y la genética, la primera concentrándose apropiadamente en las enfermedades comunes mientras que la segunda hasta hace poco utilizaba tecnología especializada para describir las raras. Los oscuros epónimos con desalentadores subtipos y términos técnicos son como pequeñas provincias de un país extranjero poco atractivo que ningún viajero global se tomará el tiempo de cartografiar. Para contrarrestar estos obstáculos es necesario un enfoque práctico de los trastornos de hipermovilidad que se centre en el reconocimiento mediante la historia clínica y el examen físico, dejando los tecnicismos de los epónimos, los subtipos y las pruebas genéticas a los subespecialistas con los que se contacta tras el primer paso vital del diagnóstico presuntivo. Una vez reconocido y derivado, el paciente flexible sigue dependiendo de su médico de cabecera, ya que sólo él puede integrar las recomendaciones y los resultados de las pruebas en el hogar médico general.
Terminología
El aumento de la amplitud de movimiento en múltiples sitios de articulación (articulación) se produce en un gran número de enfermedades, que van desde trastornos cromosómicos como el síndrome de Down hasta trastornos como el síndrome de Ehlers-Danlos (EDS), conocido por su hipermovilidad. McKusick popularizó esta categoría de trastornos conocidos por su laxitud tisular agrupándolos como trastornos hereditarios del tejido conectivo, incluido el síndrome de Marfan en el que se centró como cardiólogo. En 1977, el Dr. Peter Beighton, conocido por su escala de medición de la flexibilidad, organizó la literatura intermedia postulando 6-7 tipos de SDE, ahora simplificados por las directrices recientes como SDE clásico, vascular e hipermóvil . Ahora está claro que estos tipos tienen una considerable superposición de síntomas entre sí y con trastornos relacionados como la osteogénesis imperfecta (con numerosas fracturas), el síndrome de Marfan (dilatación/disección de grandes vasos), el síndrome de Stickler (cambios oculares y artríticos), y muchos otros.
En lugar de preocuparme por enfermedades concretas, me parece más útil centrarme en dos procesos de enfermedad principales de las displasias del tejido conectivo, ilustrados por los numerosos síntomas que se enumeran en la Tabla 1. El primero es el dolor articular por desgaste (osteoartritis), que difiere de las articulaciones hinchadas y enrojecidas de la artritis inflamatoria (autoinmune o inflamatoria) que se encuentra en la artritis reumatoide, el lupus, el síndrome de Sjogren o la espondilitis anquilosante. El segundo es el desequilibrio del sistema nervioso autónomo (disautonomía) causado por los vasos sanguíneos elásticos en los trastornos de hipermovilidad . Los vasos distensibles provocan una acumulación de sangre en la pelvis y las piernas, con una presión arterial más baja y mareos/desmayos al ponerse de pie (hipotensión ortostática). Con el tiempo, la disminución del flujo sanguíneo a la cabeza activa la rama simpática del sistema nervioso autónomo (la rama de la adrenalina «huida o lucha»), lo que provoca varios problemas: 1) el síndrome de taquicardia ortostática postural (POTS) con taquicardia, ansiedad, fatiga crónica y «niebla cerebral» (intervalos de disminución de la concentración y la memoria), 2) el síndrome del intestino irritable debido a la inhibición de la motilidad intestinal (supresión parasimpática) con estreñimiento alternado con diarrea, hinchazón, reflujo, dolor de estómago, disfagia, náuseas, y 3) trastorno de activación de mastocitos con erupciones transitorias, urticaria/piel reactiva, asma/dificultad para respirar e intolerancias a alimentos/medicamentos. He ideado un término más amplio que facilita el reconocimiento de cualquier trastorno de hipermovilidad, reconociendo las dos consecuencias principales de la laxitud articular/de los tejidos: displasia articular-autonómica o complejo articular-fatiga.
Tabla 1. Hallazgos típicos y su frecuencia en los síndromes de hipermovilidad. *De 320 pacientes femeninos y 54 masculinos mayores de 12 años evaluados mediante cuestionarios detallados en 2017; ATM, articulación temporomandibular; POTS, síndrome de taquicardia ortostática postural. Lista de síntomas adaptada en parte de Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
Historia (porcentajes)*
Infancia: Cólicos (28), dificultades de alimentación (19,4); torpe, muchas caídas (29,1)
Niño/adolescente: Aumento de peso lento (12); consciente de la flexibilidad (55,6); mostraba trucos articulares (53,6)
Dolor articular precoz (42,1); actividades del adolescente limitadas por el dolor-lesión (55,4); gafas tempranas (46,8)
Ortodoncia (61,4%) Enfermedad de las encías (11,3); esmalte pobre/muchas caries (31,6)
Problemas articulares: Chasquidos (81,6); subluxaciones (62); chasquidos, enganches, dolor en la ATM (51,2)
Dolor en al menos 2 articulaciones (80,3); cirugías articulares (33); muchos esguinces (26); fracturas >2 (31,5)
Hernia de disco vertebral (29,3)
Problemas esqueléticos: Escoliosis (31); pectus (2,2); marcha-introducción/extracción (9,3); pies planos (15,5)
Piel: Fácil aparición de hematomas (61); piel elástica (15); aterciopelada (18); cicatrices inusuales (36,3)
estrías antes del embarazo (73); cicatrización lenta (32,4)
Genitourinario (mujeres): Menorragia (61,2); endometriosis (11,3); quistes ováricos (55)
síndrome de ovario poliquístico (8,5); problemas de vejiga (46,1); hernia inguinal/femoral (11,5)
Neuromuscular: Migrañas (51,7); dolores de cabeza diarios (62,3); necesidad de medicamentos para el dolor de cabeza (45)
Deformación de Chiari (10,6); neurocirugía (2,9); entumecimiento/hormigueo (48,7); falta de equilibrio (48,6)
Dolores neuropáticos punzantes (37,3); dolores musculares/espasmos (51,7); debilidad muscular (32,4)
GI/intestinos: Estreñimiento-diarrea (67,1); hinchazón-reflujo-dolor de estómago (57,3)
Problemas de vesícula biliar (12,9); dificultad para tragar (23); náuseas frecuentes (44,9)
Corazón: Prolapso de la válvula mitral (8,7); aneurisma (0,9); arritmia distinta de la taquicardia (6,8)
POTS: Mareo al ponerse de pie (69,8); síncope (34,5); fatiga crónica (81,8); dificultades para dormir (43,5)
Niebla cerebral (mala memoria o concentración en ocasiones-70,8); sensibilidad al calor/frío (77,5); sudoración anormal (25); taquicardia (72,8); ataques de ansiedad-pánico (61,3); antojo de sal (51,2)
Immuno/mastos: Erupciones transitorias (33,4%); urticaria/piel reactiva (45,9%); asma/dificultad respiratoria (39,3); intolerancias alimentarias-medicamentos (61,5%)
Laboratorio/Imagen: Densidad ósea baja (2,1); vitamina D baja (28,7); vitamina B12 baja (2,7); hipotiroidismo (11,5); ferritina baja (1,7); hierro bajo (6,7); anticuerpos antinucleares elevados (7.1)
Número medio de hallazgos sobre 80: mujeres >12 (35,4 hallazgos) varones >12 (21,5 hallazgos)
Física (porcentaje)
Construcción: Altura >90º percentil (55,2); IMC pesado >28 (21,3); IMC delgado <19 (14,2)
Hábito marfanoide (44); aracnodactilia (46,4); signo de Walker-Murdoch presente (40,2)
Cara: Larga (25,7); cara inferior apretada (5,4); esclerótica azul-grisácea (1,6); paladar alto (53,5)
Piel: Suave (81,4); translúcida (23,8); pliegue elástico-elevado >de 1 pulgada alrededor de la mandíbula en la mitad del antebrazo (51,3)
Los pliegues consisten en la capa externa (epidérmica) (35.4); cicatrices inusuales (42,1%)
Puntuación de Beighton: 0-3 puntos (8,4); 4-6 puntos (32,6); 7-8 puntos (38,8); 9 de 9 puntos (20,2)
Otras maniobras: Juntar las manos por encima de los hombros-por detrás de la espalda (68)
Señal de oración hacia arriba por detrás de la espalda (71,3); llevar la mano alrededor de la espalda para tocar el ombligo (19,5)
Esquelético: Cifosis del cuello (41,2); escoliosis (22,3); lordosis (32,8); pies planos (39,6)
Medida de la marcha: meter o sacar los pies (28,3)
Neurológico: Debilidad muscular (11,2); disminución de la masa muscular (8,8)
Equilibrio deficiente al caminar en tándem (33,4)
Número medio de hallazgos sobre 40: mujeres >12 (17,9 hallazgos) varones >12 (15.7 hallazgos)
Displasia articular-autonómica (DAA)
Un término como «displasia articular-autonómica» o DAA puede unificar el enfoque de los trastornos de laxitud/hipermovilidad del tejido conectivo y contrarrestar tres problemas que complican su reconocimiento: 1) muchos médicos y genetistas consideran las displasias del tejido conectivo como enfermedades raras y extremas, cuando en realidad afectan a cerca del 1% de la población 2) las directrices de diagnóstico del SED, aunque simplifican los principales tipos de SED como clásico, vascular e hipermovilidad, siguen confundiendo al referirse a muchos tipos menores que forman un espectro sintomático superpuesto, y 3) la disautonomía casi obligada que acompaña al SED y a los trastornos relacionados (Tabla 1) se omite como criterio de diagnóstico, de forma análoga a describir un elefante sin mencionar su trompa. La DAA denota la inevitable conexión fisiológica entre el tejido flojo y la disautonomía, del mismo modo que los síndromes metabólicos o hepatorrenales describen cómo la obesidad conduce a la enfermedad hepática/diabetes o la insuficiencia hepática provoca la enfermedad renal.
Como la mayoría de los patrones o síndromes médicos, incluido el SED, la DAA es un complejo de síntomas con muchas causas subyacentes. «Articulo» se refiere a la articulación, reconociendo que la afección de cualquier componente articular -piel, nervio, músculo, hueso, tejido conectivo, vaso sanguíneo- causa un patrón similar de laxitud del tejido articular con desequilibrio autonómico. Habrá fragilidad de la piel, disminución de la densidad ósea con susceptibilidad a las fracturas, debilidad muscular con dolor (mialgia), neuropatía con entumecimiento/hormigueo, túnel carpiano, o síndromes de dolor regional, cambios autonómicos que incluyen taquicardia, ansiedad, sueño no reparador, inmovilidad intestinal, urticaria/explosiones, intolerancia a los alimentos y medicamentos, todo ello presente en grado variable incluso dentro de «tipos» específicos de SED. No he encontrado grandes diferencias entre los pacientes que encajan en determinados tipos de SED, como demuestran las alteraciones cutáneas más frecuentes en el SEDC clásico de tipo I frente a la mayor flexibilidad articular del SEDH hipermóvil de tipo III. Las frecuencias de los síntomas en 374 pacientes registrados por consulta sistemática no variaban significativamente entre los que diagnosticaba como cEDS frente a hEDS, e incluso los 8 pacientes identificados como posibles vEDS vasculares por tener mutaciones del gen del colágeno tipo III no eran distintivos (los aneurismas presentes en uno de los 8 pacientes tenían la misma baja frecuencia en otras categorías de EDS). Lo más importante es el concepto de un espectro de EDS, mejor representado en mi opinión como AAD.
El reconocimiento del complejo sintomático de la DAA, causado por el SED y las displasias del tejido conectivo relacionadas, evitará los tristes viajes de los pacientes de especialista en especialista, los diagnósticos sintomáticos como el vértigo, los desmayos, la fibromialgia o la fatiga crónica, y el sufrimiento trágico cuando se dispone de muchas estrategias y tratamientos preventivos. Además, el reconocimiento de la disautonomía explica la ansiedad del POTS y la depresión que frecuentemente acompaña al dolor crónico o a la limitación de la actividad , diferencias de comportamiento que pueden reconocerse como consecuencias de la enfermedad médica y no de la psiquiátrica.
Las pruebas genéticas apoyan el concepto de displasia articular-autonómica
Por su caracterización como trastornos hereditarios del tejido conectivo, se descubrió que las formas más extremas de EDS y los trastornos relacionados presentan una herencia autosómica dominante, lo que significa que los individuos afectados tienen una forma normal y otra anormal del gen (alelo). La herencia dominante tiene sentido porque la proteína defectuosa producida por el alelo anormal, análoga a un ladrillo deformado, se mezclaría con los ladrillos normales del otro alelo y formaría una estructura tambaleante. El reconocimiento de la articulación como una estructura multicomponente, constreñida por la piel suprayacente, soportada por el hueso y el cartílago interiores, y que actúa a través de nervios, músculos, tendones, revestimiento de la articulación y vasos intrínsecos, implica a su vez que cualquiera de estos elementos puede verse afectado para producir una displasia, una alteración tisular distinta de las malformaciones causadas por otros trastornos genéticos. El desarrollo de la clonación y secuenciación del ADN confirmaría el funcionamiento de la herencia autosómica dominante y la capacidad de los genes que alteran cualquier elemento de la estructura articular para causar el patrón sintomático de la DAA.
Las pruebas genéticas comenzaron centrándose en un gen «candidato» seleccionado por los síntomas del paciente, ilustrado por las pruebas del gen de la fibrilina-1 para el síndrome de Marfan . La secuenciación de las letras AGCT del ADN del gen, el «texto» de los nucleótidos, podía revelar sustituciones o «errores tipográficos» que alteraban el aminoácido de la proteína codificada y su función prevista. Luego llegó la nueva tecnología llamada NextGen o secuenciación rápida de ADN en paralelo, análoga a la lectura de todas las páginas de un libro a la vez en lugar de hacerlo de punta a punta . Esto permitió secuenciar grupos de genes (paneles) como los que causan la cardiomiopatía o la displasia del tejido conectivo o incluso los 23.000 genes de cada célula humana (nuestro genoma) en 2-3 meses . La secuenciación de las regiones genéticas que codifican proteínas o exónicas, denominada secuenciación del exoma completo (WES), puede solicitarse a muchas empresas y en mi consulta se obtuvo por menos de 200-300 dólares de coste de bolsillo en la mitad de mis pacientes (el precio de lista es de 9.000 dólares y la cantidad facturada al seguro ronda los 20.000 dólares).
Aunque la interpretación de la media de 12.000 variantes de ADN detalladas por WES sigue siendo un problema, disminuido por una epidemiología creciente de variantes en poblaciones normales y enfermas, los rendimientos de los cambios genéticos que contribuyen a los síntomas de AAD entre 620 pacientes sometidos a WES en mi práctica es más del 60%. Además de las más de 80 mutaciones de los genes del colágeno, incluidas las del colágeno de tipo I que suele asociarse a la osteogénesis imperfecta, el colágeno de tipo V que suele asociarse al SDE clásico, los colágenos de tipo VI, IX y XII descritos inicialmente con las miopatías, y el temido colágeno de tipo III asociado al SDE vascular, hay mutaciones recurrentes en los genes que afectan a los componentes cutáneos, nerviosos, musculares, fibrilares y vasculares que refuerzan un concepto de AAD en el que la alteración de cualquier componente de la articulación produce el patrón de signos y síntomas descrito en la Tabla 1. En la actualidad, el reconocimiento del SED y de los trastornos relacionados que abarca el concepto de DAA puede reforzar la impresión clínica con pruebas objetivas de ADN que permiten asesorar al paciente y a su familia.
Diagnóstico y terapia de los trastornos de hipermovilidad/AAD
La atención a los síntomas principales de dolor/fragilidad articular y autonómicos (intestino, corazón, alteración inmunológica) puede conducir a pruebas rápidas de hipermovilidad, como se muestra en la figura 1. El signo del pulgar al antebrazo puede preceder a toda la escala de Beighton, dando un punto bilateralmente para los dedos presionados hacia atrás más allá de 90 grados en la mano, tocando los pulgares a los antebrazos, hiperextendiendo el codo con los brazos extendidos, hiperextendiendo las rodillas para formar una curva de la pierna hacia atrás, y 1 punto para la capacidad de tocar las palmas al suelo . Un breve estudio de los síntomas como los de la Tabla 1 puede confirmar la sospecha, ayudado quizás por la inclusión en los exámenes físicos de algunas de las evaluaciones enumeradas en la parte inferior de la Tabla 1. Hay que tener en cuenta que no todos los pacientes con DAA son altos o delgados, y que una parte importante tendrá sobrepeso debido a las restricciones de la actividad por el dolor y la fatiga. Muchos tendrán una piel elástica, que se examina tirando hacia arriba de los pliegues alrededor de la mandíbula o en el antebrazo, especialmente cuando se trata de los pliegues finos y superficiales de la epidermis. Es frecuente la acumulación de tejido blanco o la formación de queloides alrededor de las cicatrices, y puede ser útil la evaluación de Beighton u otras maniobras de hipermovilidad enumeradas en la Tabla 1. Puede observarse una postura desplomada con curva del cuello hacia delante, escoliosis y lordosis, junto con pies planos o puntas de los pies hacia fuera (más que hacia dentro) al caminar. La fuerza muscular estará disminuida en algunos pacientes junto con un mal equilibrio al caminar en tándem, pero el generalista con tiempo limitado sólo necesita atender a algunos de estos signos físicos; la derivación establecerá el resto.
Los ortopedistas más importantes serán los que estén familiarizados con la artrosis y los cambios que la acompañan en el SED, como los cambios degenerativos de las articulaciones, el deterioro del cartílago, las bandas de plica y las regiones de hueso muerto. Es deseable un ortopedista experimentado y conservador que reconozca los malos resultados de las cirugías para «apretar» o reemplazar las articulaciones. Un cardiólogo puede documentar el POTS mediante la prueba de la mesa basculante para detectar cambios ortostáticos en la frecuencia cardíaca y la presión arterial, realizando también estudios electro y ecocardiográficos para excluir la dilatación aórtica o los aneurismas. La neurología puede evaluar las migrañas comunes o las cefaleas diarias, la deformación de Chiari que se produce con el deslizamiento y la herniación del tejido cerebral laxo, la neuropatía debida a la constricción dentro de las articulaciones laxas y los cambios musculares denotados por la descripción de los síntomas de la fibromialgia y resaltados por los frecuentes cambios genéticos miopáticos que están fomentando un nuevo término de EDS miopático. Los alergólogos familiarizados con la activación de los mastocitos y los gastroenterólogos que pueden documentar la baja motilidad intestinal son adiciones valiosas al equipo de la subespecialidad, y nadie es más valioso que el médico de cabecera para gestionar las numerosas recomendaciones y medicamentos que pueden ser necesarios. Aunque pueda parecer extraño viniendo de un genetista, creo que las evaluaciones de la subespecialidad que conducen a la terapia son más importantes que la evaluación y las pruebas genéticas, pero estas últimas pueden ser útiles para desactivar las suposiciones sobre la enfermedad psiquiátrica.
El tratamiento comienza con un programa de ejercicios que puede beneficiar tanto a la artritis debida a la hipermovilidad articular como a su disautonomía refleja. La atención al dolor articular (los dolores de crecimiento se producen por la noche) o a las lesiones/deformidades esqueléticas puede instituir la prevención haciendo que el paciente sea consciente de su condición (nótese en la Tabla 1 que muchos no reconocen su flexibilidad como anormal). Las lesiones por desgaste pueden prevenirse colocando fajas en las articulaciones susceptibles, bandas en la cintura para apoyar la espalda y eligiendo actividades que protejan las articulaciones, como nadar en lugar de correr. Los pacientes deben mantenerse activos levantando pesas con moderación y realizando otras actividades razonables para fortalecer los músculos alrededor de las articulaciones, evitando los ciclos de inactividad que exacerban la fatiga crónica o la fibromialgia. La fisioterapia adecuada, el control del dolor, la orientación del ejercicio y los medicamentos anecdóticos como los preparados de glucosamina/condroitina han ayudado a muchos, pero deben evitarse los relajantes que quitan la protección muscular de las articulaciones y las inyecciones de esteroides que enmascaran las lesiones, promueven la osteoporosis/degeneración y sólo dan un alivio temporal.
El pilar de la terapia de la disautonomía es la hidratación (8-10 vasos de líquido al día), la sal (en ausencia de hipertensión lábil) y una dieta rica en proteínas, todo ello dirigido a aumentar el volumen de líquido sanguíneo y mejorar la circulación cerebral. La terapia nutricional adicional puede incluir vitamina C (2 g al día) vitamina D (>2000 unidades al día, más que las 400-800 de las multivitaminas), vitamina B12 (1-2,5 mg al día). La baja motilidad intestinal o gastroparesia se favorece evitando los líquidos antes de las comidas, comidas más pequeñas y frecuentes, probióticos, preparados enzimáticos y, según informan muchos, disminución del gluten y los lácteos que son irritantes comunes. Los programas de ejercicio en posición supina, la biorretroalimentación y muchas opciones de medicación están disponibles a través de cardiólogos para el POTS (por ejemplo, betabloqueantes, Midodrine, Florinef), a través de neurólogos o expertos en el manejo del dolor (por ejemplo, Gabapentin, Lyrica), o a través de alergólogos que pueden proporcionar protocolos de antihistamínicos para suprimir los mastocitos (por ejemplo, Zyrtec, Zantac, Cromolyn, Singulair). Las ventajas de las pruebas genómicas, como el WES, que van más allá de los genes conocidos como causantes de la displasia del tejido conectivo, son los nuevos hallazgos, como los genes relacionados con el músculo que ahora se sabe que causan el SED, y la documentación de hallazgos incidentales o secundarios, como las mutaciones genéticas del cáncer de mama-ovario. El hallazgo de genes asociados a síntomas más graves permite a los pacientes conocer los riesgos para la descendencia y elegir estrategias de diagnóstico genético prenatal o preimplantacional compatibles con sus valores.
Presentación del caso
Eve acudió a su médico a los 18 años, mareada, desmayada, ansiosa y con náuseas, tan agotada que se retiraba los viernes por la noche para descansar todo el fin de semana. Había gozado de buena salud, participaba activamente en la gimnasia y la animación, era una chica valiente y brillante cuya perseverancia a pesar de las lesiones se ganaba los aplausos de sus entrenadores. Luego, una gripe de la que nunca se recuperó, náuseas tan intensas que no podía comer, pérdida de peso, desmayos en clase, despertares nocturnos con el corazón palpitante, despertares matutinos sin sueño reparador. Eve acudió a muchos médicos, incluso a urgencias cuando las palpitaciones y el pánico la dejaron sin aliento. No había nada malo, excepto el estrés, decían, todo estaba en su mente. Calmada pero no curada por las pastillas, el trabajo fallaba por la fatiga y la confusión, y se preguntaba si se estaba volviendo loca. No se reconocía su piel suave y frágil, sus menstruaciones duras y abundantes, sus dolores de cabeza diarios y, lo más llamativo de todo, una hipermovilidad que ella y su familia asumían como normal: «Sí, somos doblados y con doble articulación, ¿no lo somos todos?». Eve habría respondido si le hubieran preguntado.
Afortunadamente, Eve buscó un nuevo médico de cabecera que pudo documentar muchos de los signos y síntomas enumerados en la Tabla I. Eve tenía cólicos tempranos y reflujo con dificultad para amamantar, era muy flexible, su madre la llamaba la bebé «sin huesos», y cuando era mayor se sentaba sobre las rodillas con las piernas extendidas detrás de ella (la posición «W»), diciendo que era más cómodo. Al principio era lenta para caminar, se caía con frecuencia y parecía torpe, pero pronto se mostró activa y trepaba por todas partes, haciendo que sus padres se estremecieran al ponerle un pie alrededor del cuello o al hacer los splits con facilidad. Pero al entrar en la escuela primaria empezó a quejarse de artralgias, le dolían las piernas por la noche y le molestaban las rodillas y los tobillos después de la actividad. Su pediatra tranquilizó a sus padres diciéndoles que sólo tenía «dolores de crecimiento», pero se preocuparon más cuando intentó jugar al fútbol y siguió dándose la vuelta a los tobillos, un esguince que requirió una bota durante 2 meses después de que se observara una fractura por estrés.
Eve tuvo que dejar el fútbol, pero descubrió que sobresalía en la gimnasia a medida que crecía, eligiendo los ejercicios de suelo debido a su escaso equilibrio en la viga y a la tendencia a que se le saliera el hombro en las barras paralelas. Se dio cuenta de que sus articulaciones estallaban con el movimiento y a menudo las hacía estallar a propósito porque parecía aliviar la tensión y el dolor. Probó a ser animadora con éxito en las maniobras, pero descubrió que era demasiado sensible al calor. Las otras chicas la llamaban «cara de tomate» porque se sobrecalentaba durante las rutinas, y se cansaba fácilmente, necesitando a menudo un día de descanso después del ejercicio. Sus problemas digestivos reaparecieron con frecuentes estreñimientos y dolores de estómago, y a menudo se sentía hinchada y necesitaba laxantes. En 6º curso faltó a la escuela por sus problemas intestinales y su dolor de articulaciones, y a menudo se sentía fatigada y mareada. No se sentía descansada por las mañanas y a menudo echaba la siesta los fines de semana, lo que suponía un cambio en su anterior estilo de vida activo y entusiasta. A menudo se sentía ansiosa, sin poder atribuirlo a ningún estrés escolar o familiar específico, y empezó a tener dolores de cabeza por la mañana que creía que se debían a su mal sueño. Sus padres volvieron a llevarla al pediatra y, una vez más, les dijeron que no había nada malo, sino el estrés escolar habitual.
Cuando tenía 14 años, Eve tuvo una grave enfermedad parecida a la gripe y faltó a clase durante un mes con una gran fatiga, dolores de cabeza, náuseas, vómitos y pérdida de peso. Tenía terribles mareos y algo de dolor en el pecho, sintiendo que su corazón latía con fuerza a veces (tenía palpitaciones). Sus padres pidieron consejo a otro pediatra y éste envió a Eve a un cardiólogo, que pensó que tenía taquicardia por el estrés y la ansiedad que rodeaban su enfermedad. Le recetó propranolol, un betabloqueante, para reducir el ritmo cardíaco, pero la niña se fatigó aún más y pasó gran parte del día en la cama. Sus dolores articulares aumentaron tras la enfermedad y permanecía en la cama la mayor parte del día, sintiéndose triste y poniéndose nerviosa cuando sus padres intentaban sacarla al exterior o llevarla a dar un paseo. La llevaron de nuevo al segundo pediatra, que la derivó a psiquiatría, y se empezó a administrar Zoloft para ayudarla con la ansiedad y la depresión. A los pocos días a Eve le salió una erupción con urticaria y tuvo que dejar esa medicación. Siguió teniendo mareos y la vieron dos especialistas más, un gastroenterólogo que pensó que podía ser celíaca y un otorrinolaringólogo que le diagnosticó vértigo y la envió a neurología. Ella le dijo a ese médico que tenía dolores de cabeza que emanaban de la parte posterior de su cabeza, y se obtuvo un estudio de resonancia magnética de la cabeza en posición supina que fue normal.
La apreciación del probable diagnóstico de DAA por parte de su médico de cabecera instituyó una ronda diferente de remisiones a subespecialidades, incluyendo neurología, donde un estudio de resonancia magnética de la cabeza en posición vertical demostró la herniación de la deformación de Chiari. Un cardiólogo documentó una prueba de mesa basculante anormal junto con una baja motilidad intestinal y demostró que su ansiedad y estrés-fatiga se debían a la disautonomía. Las estrategias de hidratación nutricional, sal y proteínas, junto con el tratamiento con betabloqueantes y antihistamínicos, mejoraron su fatiga y sus síntomas intestinales y disminuyeron sus dolores de cabeza. Se decidió que la hernia de Chiari de 4-5 mm no era suficiente para requerir una intervención quirúrgica, y se reconoció que su tratamiento psiquiátrico era un tratamiento de los síntomas secundarios de un trastorno médico. Algunos de sus subespecialistas, escépticos ante su diagnóstico clínico, se convencieron gracias a las pruebas genéticas que documentaron un cambio en un gen de la cadena alfa-1 del colágeno tipo V, abreviado COL5A1, un hallazgo común en los pacientes con SED. Eve había heredado la mutación de su madre, que había tenido síntomas más leves de TEA y ahora era capaz de controlarlos mejor.
A lo largo de varios meses, la salud y la vitalidad de Eve se restablecieron y «recuperó su vida», como dirían ella y sus padres. Seguía teniendo dolor en las articulaciones, ansiedad y fatiga en ocasiones, pero el «ejercicio sabio» para crear protección muscular alrededor de sus articulaciones, los enfoques nutricionales para la disautonomía y los enfoques de biorretroalimentación para evitar la ansiedad le cambiaron la vida. Todos estos tratamientos se debieron al reconocimiento de su médico de cabecera, que ahora puede coordinar la atención a Eve y a su madre y proporcionar recursos para comprender tanto el SED como sus cambios genéticos subyacentes. La transición a la siguiente generación fue la capacidad de informar a Eve de su riesgo del 50% de transmitir su mutación COL5A y algunos riesgos de parto prematuro y hemorragia posparto en cualquier embarazo futuro .
- Remvig L, Jensen DV, Ward RC (2007) Epidemiología de la hipermovilidad articular general y base de los criterios propuestos para el síndrome de hipermovilidad articular benigna: revisión de la literatura. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. En: Connective Tissue and its Heritable Disorders. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Disautonomía en el síndrome de hipermovilidad articular. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lessons in pain relief–a personal postgraduate experience. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) Una pequeña molécula para una gran enfermedad. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Capítulo 11: Secuenciación de genes y genomas: Interpretación de la variación genética a nivel de nucleótidos. En: Human Chromosome Variation: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing–are we there yet? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Reescribiendo la historia natural del dolor y los síntomas relacionados en el síndrome de hipermovilidad articular/síndrome de Ehlers-Danlos, tipo hipermovilidad. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Síndrome de hipermovilidad articular benigna: evaluación, diagnóstico y manejo. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Molestias musculoesqueléticas asociadas a la hipermovilidad articular generalizada. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Diagnóstico genético presintomático y preimplantacional: Neurología, NextGenetics y la próxima generación. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Manejo del dolor y la fatiga en el síndrome de hipermovilidad articular (también conocido como síndrome de Ehlers- Danlos, tipo hipermovilidad): principios y propuesta de un enfoque multidisciplinar. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Reescribiendo la historia natural del dolor y los síntomas relacionados en el síndrome de hipermovilidad articular/síndrome de Ehlers-Danlos, tipo hipermovilidad. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Tratamiento multidisciplinar de la discapacidad en el síndrome de Ehlers-Danlos tipo hipermovilidad/síndrome de hipermovilidad: un estudio piloto utilizando una combinación de terapia física y cognitivo-conductual en 12 mujeres. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Análisis del exoma de la displasia del tejido conectivo: ¿muerte y renacimiento de la genética clínica? Am J Med Genet A 164A: 1209-1212.