Mots-clés
hypermobilité, dysplasie du tissu conjonctif, syndrome d’ehlers-danlos, échelle de beighton, dysautonomie, syndrome de tachycardie orthostatique posturale, syndrome du côlon irritable, trouble de l’activation des mastocytes, dysplasie articulo-autonomique
Introduction
Très peu reconnus et tragiquement maltraités sont les 20% de femmes et 10% d’hommes dont les tissus sont plus souples et plus fragiles que la moyenne . Peu de possibilités de prévention et de traitement dépassent celle de reconnaître l’hypermobilité, facilement dépistée en demandant au patient de pousser son pouce contre son avant-bras (figure 1). Ce signe révélateur peut ouvrir la voie à une évaluation plus détaillée qui est tout à fait dans les cordes du médecin généraliste, dont les vastes connaissances cliniques correspondent bien aux problèmes multisystémiques qui peuvent découler d’un relâchement du tissu conjonctif. Je passe ici en revue la nature des troubles de l’hypermobilité, je démontre que des connaissances générales plutôt que de sous-spécialité peuvent conduire à des thérapies qui changent la vie de ces patients, et je montre comment les tests génétiques font entrer des descriptions symptomatiques comme la fibromyalgie ou le syndrome de fatigue chronique dans la lumière perspicace de la médecine de précision.
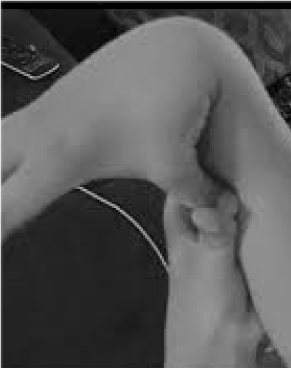
Figure 1. Hypermobilité.
Plusieurs obstacles doivent être surmontés avant que les patients souffrant d’hypermobilité puissent recueillir les avantages de la médecine générale. La première provient de l’orientation pionnière mais myope des dermatologues Ehlers et Danlos imprimant un étirement incroyable et exotique de la peau comme symbole trompeur des troubles de la laxité du tissu conjonctif. Un deuxième obstacle est le fossé croissant entre la médecine pratique et la médecine génétique, la première se concentrant à juste titre sur les maladies courantes tandis que la seconde, jusqu’à récemment, utilisait des technologies spécialisées pour décrire les maladies rares. Les éponymes obscurs, les sous-types déconcertants et les termes techniques sont comme les petites provinces d’un pays étranger peu attrayant qu’aucun voyageur mondial ne prendra le temps de cartographier. Pour contrer ces obstacles, il faut une approche pratique des troubles de l’hypermobilité qui se concentre sur la reconnaissance par l’anamnèse et l’examen physique, laissant les détails techniques des éponymes, sous-types et tests génétiques aux sous-spécialistes contactés après la première étape vitale du diagnostic présumé. Une fois reconnu et référé, le patient flexible reste dépendant de son médecin traitant, car lui seul peut intégrer les recommandations et les résultats des tests dans le foyer médical général.
Terminologie
Une augmentation de l’amplitude des mouvements au niveau de plusieurs articulations (joints) se produit dans un grand nombre de maladies, allant de troubles chromosomiques comme le syndrome de Down à des troubles comme le syndrome d’Ehlers-Danlos (SED) connus pour leur hypermobilité. McKusick a popularisé cette catégorie de troubles connus pour leur laxité tissulaire en les regroupant sous le nom de troubles héréditaires du tissu conjonctif, dont le syndrome de Marfan sur lequel il s’est concentré en tant que cardiologue. En 1977, le Dr Peter Beighton, connu pour son échelle de mesure de la flexibilité, a organisé la littérature intermédiaire en postulant 6-7 types de SDE, maintenant simplifiés par les directives récentes comme SDE classique, vasculaire et hypermobile . Il est maintenant clair que ces types ont un chevauchement considérable de symptômes entre eux et des troubles apparentés comme l’ostéogenèse imparfaite (avec de nombreuses fractures), le syndrome de Marfan (dilatation/dissection des gros vaisseaux), le syndrome de Stickler (changements oculaires et arthritiques), et bien d’autres.
Plutôt que de s’intéresser à des maladies particulières, je trouve plus utile de me concentrer sur deux processus pathologiques majeurs des dysplasies du tissu conjonctif, illustrés par les nombreux symptômes énumérés dans le tableau 1. La première est la douleur articulaire due à l’usure (arthrose) qui diffère des articulations rouges et gonflées de l’arthrite inflammatoire (auto-immune ou inflammatoire) rencontrée dans la polyarthrite rhumatoïde, le lupus, le syndrome de Sjögren ou la spondylarthrite ankylosante. Le second est le déséquilibre du système nerveux autonome (dysautonomie) causé par l’élasticité des vaisseaux sanguins dans les troubles de l’hypermobilité . Les vaisseaux distendus provoquent une accumulation de sang dans le bassin et les jambes, avec une baisse de la tension artérielle et des vertiges/une sensation de faiblesse en position debout (hypotension orthostatique). Avec le temps, la diminution du flux sanguin vers la tête active le bras sympathique du système nerveux autonome (le bras « fuite ou combat » de l’adrénaline), ce qui entraîne plusieurs problèmes : 1) le syndrome de tachycardie orthostatique posturale (POTS) avec tachycardie, anxiété, fatigue chronique et « brouillard cérébral » (intervalles de baisse de concentration et de mémoire), 2) le syndrome du côlon irritable dû à l’inhibition de la motilité intestinale (suppression parasympathique) avec constipation alternant avec diarrhée, ballonnements, reflux, douleurs d’estomac, dysphagie, nausées, et 3) trouble de l’activation des mastocytes avec éruptions cutanées transitoires, urticaire/peau réactive, asthme/essoufflement, et intolérances alimentaires/médicamenteuses. J’ai imaginé un terme plus large qui facilite la reconnaissance de tout trouble d’hypermobilité, reconnaissant les deux principales conséquences de la laxité articulaire/tissulaire : la dysplasie articulo-autonomique ou le complexe articulation-fatigue.
Tableau 1. Résultats typiques et leur fréquence dans les syndromes d’hypermobilité. *Sur 320 patientes et 54 patients âgés de plus de 12 ans évalués par des questionnaires détaillés en 2017 ; ATM, articulation temporomandibulaire ; POTS, syndrome de tachycardie orthostatique posturale. Liste de symptômes adaptée en partie de Wilson GN, Cooley WC : Preventive Health Care for Children with Genetic Condition : Providing a Medical Home. 2e éd. Cambridge, MA. Cambridge University Press, 2006.
HISTOIRE (pourcentages)*
Infantile : Coliques (28), difficultés d’alimentation (19,4) ; maladroit, nombreuses chutes (29,1)
Enfant/adolescent : Prise de poids lente (12) ; conscient de la flexibilité (55,6) ; a montré des astuces pour les articulations (53,6)
Douleurs articulaires précoces (42,1) ; activités de l’adolescent limitées par la douleur-blessure (55,4) ; lunettes précoces (46,8)
orthodontie (61,4%) Maladie des gencives (11,3) ; mauvais émail/nombreuses caries (31,6)
Problèmes articulaires : Craquements (81,6) ; subluxations (62) ; ATM craquements, accrochages, douleurs (51,2)
Douleurs dans au moins 2 articulations (80,3) ; chirurgies articulaires (33) ; nombreuses entorses (26) ; fractures >2 (31,5)
Hernie discale vertébrale (29,3)
Problèmes squelettiques : Scoliose (31) ; pectus (2,2) ; démarche – pied dedans/dehors (9,3) ; pieds plats (15,5)
Peau : Ecchymoses faciles (61) ; peau extensible (15) ; veloutée (18) ; cicatrices inhabituelles (36,3)
stries avant la grossesse (73) ; cicatrisation lente (32,4)
Génito-urinaire (femmes) : Ménorragie (61,2) ; endométriose (11,3) ; kystes ovariens (55)
syndrome des ovaires polykystiques (8,5) ; problèmes de vessie (46,1) ; hernie inguinale/fémorale (11,5)
Neuromusculaire : Migraines (51,7) ; maux de tête quotidiens (62,3) ; besoin d’un médicament contre les maux de tête (45)
Déformation de Chiari (10,6) ; neurochirurgie (2,9) ; engourdissement/troubles (48,7) ; mauvais équilibre (48,6)
Douleurs fulgurantes neuropathiques (37,3) ; douleurs musculaires/spasmes (51,7) ; faiblesse musculaire (32,4)
GI/intestinaux : Constipation-diarrhée (67,1) ; ballonnements-reflux-douleurs d’estomac (57,3)
Problèmes de vésicule biliaire (12,9) ; déglutition difficile (23) ; nausées fréquentes (44,9)
Cœur : Prolapsus de la valve mitrale (8,7) ; anévrisme (0,9) ; arythmie autre que tachycardie (6,8)
POTS : Étourdissements en position debout (69,8) ; syncope (34,5) ; fatigue chronique (81,8) ; difficultés de sommeil (43,5)
Brouillard cérébral (mauvaise mémoire ou concentration par moments-70,8) ; sensibilité à la chaleur/froid (77,5) ; transpiration anormale (25) ; tachycardie (72,8) ; anxiété/attaques de panique (61,3) ; envie de sel (51,2)
Immuno/masto : Éruptions cutanées transitoires (33,4 %) ; urticaire/peau réactive (45,9 %) ; asthme/essoufflement (39,3) ; intolérances aux aliments/médicaments (61,5 %)
Labos/imagerie : Faible densité osseuse (2,1) ; faible taux de vitamine D (28,7) ; faible taux de vitamine B12 (2,7) ; hypothyroïdie (11,5) ; faible taux de ferritine (1,7) ; faible taux de fer (6,7) ; anticorps antinucléaires élevés (7.1)
Nombre moyen de découvertes sur 80 : femmes >12 (35,4 découvertes) hommes >12 (21,5 découvertes)
Physique (pourcentage)
Construction : Grande taille >90e percentile (55,2) ; lourd-BMI >28 (21,3) ; mince-BMI <19 (14,2)
Habitude marfanoïde (44) ; arachnodactylie (46,4) ; signe de Walker-Murdoch présent (40,2)
Face : Long (25,7) ; face inférieure serrée (5,4) ; sclérotique bleu-gris (1,6) ; palais haut (53,5)
Peau : Douce (81,4) ; translucide (23,8) ; soulèvement élastique >Plis de 1 pouce autour de la mâchoire sur le milieu de l’avant-bras (51,3)
Les plis sont constitués de la couche externe (épidermique) (35.4) ; cicatrices inhabituelles (42,1%)
Score de Beighton : 0-3 points (8,4) ; 4-6 points (32,6) ; 7-8 points (38,8) ; 9 de 9 points (20,2)
Autres manœuvres : Joindre les mains sur l’épaule-derrière le dos (68)
Signe de prière vers le haut derrière le dos (71,3) ; amener la main autour du dos pour toucher l’ombilic (19,5)
Squelettique : Cyphose du cou (41,2) ; scoliose (22,3) ; lordose (32,8) ; pieds plats (39,6)
Marche – pied en dedans ou en dehors (28,3)
Névrologique : Faiblesse musculaire (11,2) ; diminution de la masse musculaire (8,8)
Mauvais équilibre par marche en tandem (33,4)
Nombre moyen de constatations sur 40 : femmes >12 (17,9 constatations) hommes >12 (15.7 découvertes)
Dysplasie articulo-autonomique (DAA)
Un terme comme « dysplasie articulo-autonomique » ou DAA peut unifier l’approche des troubles de la laxité/hypermobilité du tissu conjonctif et contrer trois problèmes qui compliquent leur reconnaissance : 1) de nombreux médecins et généticiens considèrent les dysplasies du tissu conjonctif comme des maladies rares et extrêmes, alors qu’elles touchent en fait environ 1 % de la population ; 2) les directives de diagnostic du SDE, bien qu’elles simplifient les principaux types de SDE (classique, vasculaire et hypermobilité), prêtent encore à confusion en faisant référence à de nombreux types mineurs qui forment un spectre de symptômes qui se chevauchent ; et 3) la dysautonomie presque obligatoire qui accompagne le SDE et les troubles apparentés (tableau 1) est omise en tant que critère de diagnostic, comme si l’on décrivait un éléphant sans mentionner sa trompe. L’AAD désigne l’inévitable connecteur physiologique entre le tissu lâche et la dysautonomie de la même manière que les syndromes métaboliques ou hépatorénaux décrivent comment l’obésité entraîne une maladie du foie/diabète ou comment l’insuffisance hépatique entraîne une maladie rénale.
Comme la plupart des schémas ou syndromes médicaux, y compris le SDE, l’AAD est un complexe de symptômes avec de nombreuses causes sous-jacentes. « Articulo » fait référence à l’articulation, en reconnaissant que l’affliction de n’importe quel composant articulaire – peau, nerf, muscle, os, tissu conjonctif, vaisseau sanguin – provoque un schéma similaire de laxité des tissus articulaires avec un déséquilibre autonome. Il y aura une fragilité de la peau, une diminution de la densité osseuse avec une susceptibilité aux fractures, une faiblesse musculaire avec des douleurs (myalgie), une neuropathie avec des engourdissements et des picotements, un canal carpien ou des syndromes de douleur régionale, des changements autonomes, y compris la tachycardie, l’anxiété, un sommeil non réparateur, une immotilité intestinale, de l’urticaire et des éruptions cutanées, des intolérances alimentaires et médicamenteuses, tous présents à des degrés variables, même dans des « types » spécifiques de SDE. Je n’ai pas trouvé de grandes différences entre les patients qui correspondent à des types particuliers de SDE, illustrées par des changements cutanés plus fréquents dans le cEDS classique de type I contre une plus grande flexibilité articulaire dans le hEDS hypermobile de type III. Les fréquences des symptômes chez 374 patients enregistrés par interrogation systématique ne variaient pas de manière significative entre ceux que j’ai diagnostiqués comme cEDS ou hEDS, et même les 8 patients identifiés comme vEDS vasculaire possible en raison de mutations du gène du collagène de type III ne se distinguaient pas (les anévrismes présents chez l’un des 8 patients avaient la même faible fréquence dans les autres catégories d’EDS). Le plus important est le concept d’un spectre de SDE, mieux représenté à mon avis comme AAD.
La reconnaissance du complexe de symptômes AAD, causé par le SDE et les dysplasies du tissu conjonctif associées, évitera aux patients de tristes voyages de spécialiste en spécialiste, des diagnostics symptomatiques comme le vertige, l’évanouissement, la fibromyalgie ou la fatigue chronique, et des souffrances tragiques alors que de nombreuses stratégies préventives et traitements sont disponibles. En outre, la reconnaissance de la dysautonomie explique l’anxiété du POTS et la dépression qui accompagne fréquemment la douleur chronique ou la limitation d’activité , des différences de comportement qui peuvent être reconnues comme des conséquences d’une maladie médicale plutôt que psychiatrique.
Les tests génétiques soutiennent le concept de dysplasie articulo-autonomique
Impliquées par leur caractérisation comme des troubles héréditaires du tissu conjonctif , les formes les plus extrêmes du SDE et des troubles apparentés se sont avérées présenter une hérédité autosomique dominante, ce qui signifie que les individus affectés ont une forme normale et une forme anormale du gène (allèle). L’hérédité dominante est logique car la protéine défectueuse produite par l’allèle anormal, analogue à une brique déformée, se mélangerait aux briques normales de l’autre allèle et formerait une structure bancale. La reconnaissance de l’articulation en tant que structure à plusieurs composants, limitée par la peau sus-jacente, soutenue par l’os et le cartilage internes, et agissant par l’intermédiaire de nerfs, de muscles, de tendons, d’un revêtement articulaire et de vaisseaux intrinsèques, implique à son tour que n’importe lequel de ces éléments peut être affecté et produire une dysplasie, une perturbation tissulaire distincte des malformations causées par d’autres troubles génétiques. Le développement du clonage et du séquençage de l’ADN confirmerait le fonctionnement de l’hérédité autosomique dominante et la capacité des gènes altérant n’importe quel élément de la structure articulaire à provoquer le schéma symptomatique de l’AAD.
Les tests génétiques ont commencé par se concentrer sur un gène « candidat » sélectionné en raison des symptômes du patient, illustré par le test du gène de la fibrilline-1 pour le syndrome de Marfan . Le séquençage des lettres AGCT de l’ADN du gène, le « texte » des nucléotides, pouvait révéler des substitutions ou des « typos » qui modifiaient l’acide aminé de la protéine codée et sa fonction prévue. Puis est apparue la nouvelle technologie appelée NextGen ou séquençage parallèle rapide de l’ADN, analogue à la lecture de toutes les pages d’un livre en une seule fois plutôt que de bout en bout . Cette technologie a permis de séquencer des groupes de gènes (panels), tels que ceux responsables de la cardiomyopathie ou de la dysplasie du tissu conjonctif, voire l’ensemble des 23 000 gènes de chaque cellule humaine (notre génome) en 2 ou 3 mois. Le séquençage des régions génétiques codant pour les protéines ou exoniques, appelé séquençage de l’exome entier (WES), peut être commandé auprès de nombreuses sociétés et, dans ma pratique, a été obtenu pour moins de 200 à 300 dollars de frais à la charge de la moitié de mes patients (le prix catalogue est de 9000 dollars et le montant facturé à l’assurance est d’environ 20 000 dollars).
Bien que l’interprétation des 12 000 variants d’ADN en moyenne détaillés par le WES reste un problème, diminué par une épidémiologie croissante des variants dans les populations normales et malades, les rendements des changements de gènes contribuant aux symptômes de l’AAD parmi 620 patients subissant un WES dans ma pratique est de plus de 60%. En plus de plus de 80 mutations des gènes du collagène, y compris celles du collagène de type I généralement associé à l’ostéogenèse imparfaite, du collagène de type V souvent associé au SDE classique, des collagènes de type VI, IX et XII initialement décrits avec les myopathies, et du collagène de type III redouté associé au SDE vasculaire, il existe des mutations récurrentes dans les gènes affectant les composants de la peau, des nerfs, des muscles, des fibrilles et des vaisseaux qui renforcent un concept de DAA où l’altération de n’importe quel composant articulaire produit le schéma de signes et de symptômes décrit dans le tableau 1. Désormais, la reconnaissance du SDE et des troubles apparentés englobés dans le concept de DAA peut étayer l’impression clinique avec des preuves objectives d’ADN qui permettent de conseiller le patient et sa famille.
Diagnostic et thérapie des troubles d’hypermobilité/DAA
La vigilance à l’égard des principaux symptômes de douleur/fragilité articulaire et de perturbation autonome (intestinale, cardiaque, immunitaire) peut conduire à des tests rapides d’hypermobilité comme le montre la figure 1. Le signe du pouce sur l’avant-bras peut préfigurer toute l’échelle de Beighton, donnant un point bilatéralement pour les doigts repoussés au-delà de 90 degrés sur la main, le contact des pouces sur les avant-bras, l’hyperextension du coude avec les bras étendus, l’hyperextension des genoux pour former une courbe de la jambe vers l’arrière, et 1 point pour la capacité de toucher les paumes sur le sol. Un bref examen des symptômes tels que ceux du tableau 1 peut confirmer les soupçons, en incluant peut-être dans les examens physiques certaines des évaluations énumérées dans la partie inférieure du tableau 1. Notez que tous les patients atteints de DAA ne sont pas grands ou minces, et qu’une grande partie d’entre eux seront en surpoids en raison des restrictions d’activité dues à la douleur et à la fatigue. Beaucoup auront une peau élastique, examinée en tirant sur les plis autour de la mâchoire ou sur l’avant-bras, en particulier lorsqu’il s’agit des plis fins et superficiels de l’épiderme. L’accumulation de tissu blanc ou la formation de chéloïdes autour des cicatrices sont fréquentes, et l’évaluation de Beighton ou d’autres manœuvres d’hypermobilité énumérées dans le tableau 1 peuvent être utiles. On peut noter une posture avachie avec une courbure du cou vers l’avant, une scoliose et une lordose, ainsi que des pieds plats ou des orteils en dehors (plus qu’en dedans) lors de la marche. La force musculaire sera en baisse chez certains patients ainsi qu’un mauvais équilibre lors de la marche en tandem, mais le généraliste disposant d’un temps limité ne doit s’occuper que de quelques-uns de ces signes physiques ; la référence établira le reste.
Les orthopédistes les plus importants seront ceux qui connaissent bien l’arthrose et les changements qui l’accompagnent dans les SDE comme les changements dégénératifs des articulations, la détérioration du cartilage, les bandes de plica et les régions d’os mort. Il est souhaitable d’avoir un orthopédiste expérimenté et conservateur qui reconnaît les mauvais résultats des interventions chirurgicales visant à « resserrer » ou à remplacer les articulations. Un cardiologue peut documenter le SOPT en utilisant le test de la table basculante pour les changements orthostatiques de la fréquence cardiaque et de la pression artérielle, et en effectuant également des études électro- et échocardiographiques pour exclure une dilatation de l’aorte ou des anévrismes. La neurologie peut évaluer les migraines courantes ou les maux de tête quotidiens, la déformation de Chiari qui se produit avec le glissement et l’hernie des tissus cérébraux laxatifs, la neuropathie due à la constriction dans les articulations laxatives, et les changements musculaires dénotés par la description des symptômes de la fibromyalgie et mis en évidence par les changements fréquents des gènes myopathiques qui favorisent un nouveau terme de SDE myopathique. Les allergologues qui connaissent l’activation des mastocytes et les gastro-entérologues qui peuvent documenter une faible motilité intestinale sont des ajouts précieux à l’équipe de la sous-spécialité, et personne n’est plus précieux que le médecin traitant pour gérer les nombreuses recommandations et les médicaments qui peuvent être nécessaires. Bien que cela puisse sembler étrange venant d’un généticien, je pense que les évaluations des sous-spécialités qui mènent à la thérapie sont plus importantes que l’évaluation et les tests génétiques, mais ces derniers peuvent être utiles pour désamorcer les hypothèses sur la maladie psychiatrique .
Le traitement commence par un programme d’exercices qui peut bénéficier à la fois à l’arthrite due à l’hypermobilité articulaire et à sa dysautonomie réflexe. L’attention portée aux douleurs articulaires (les douleurs de croissance surviennent la nuit) ou aux blessures/déformations squelettiques peut instituer une prévention en faisant prendre conscience au patient de son état (notez dans le tableau 1 que beaucoup ne reconnaissent pas leur flexibilité comme anormale). Les blessures dues à l’usure peuvent être évitées en renforçant les articulations sensibles, en utilisant des bandes de taille pour soutenir le dos et en choisissant des activités qui protègent les articulations comme la natation plutôt que le jogging. Les patients doivent rester actifs en faisant de la musculation modérée et d’autres activités raisonnables pour renforcer les muscles autour des articulations, afin d’éviter les cycles d’inactivité qui exacerbent la fatigue chronique ou la fibromyalgie. La thérapie physique appropriée, la gestion de la douleur, les conseils en matière d’exercice et les médicaments anecdotiques comme les préparations à base de glucosamine/chondroïtine ont aidé beaucoup de gens, mais il faut éviter les relaxants qui enlèvent la protection musculaire des articulations et les injections de stéroïdes qui masquent les blessures, favorisent l’ostéoporose/dégénérescence et ne donnent qu’un soulagement temporaire.
Le pilier de la thérapie de la dysautonomie est l’hydratation (8-10 verres de liquide par jour), le sel (hypertension labile absente), et un régime riche en protéines, le tout visant à augmenter le volume de liquide sanguin et à améliorer la circulation cérébrale. Une thérapie nutritionnelle supplémentaire peut inclure la vitamine C (2 g par jour), la vitamine D (>2000 unités par jour, plus que les 400-800 des multivitamines), la vitamine B12 (1-2,5 mg par jour). La faible motilité intestinale ou la gastroparésie est améliorée en évitant les liquides avant les repas, en prenant des repas plus petits et plus fréquents, en prenant des probiotiques, des préparations enzymatiques et, selon de nombreux témoignages, en réduisant le gluten et les produits laitiers qui sont des irritants courants. Des programmes d’exercices en position couchée, le biofeedback et de nombreux médicaments sont disponibles auprès des cardiologues pour le POTS (par exemple, les bêta-bloquants, Midodrine, Florinef), auprès des neurologues ou des experts en gestion de la douleur (par exemple, Gabapentin, Lyrica), ou auprès des allergologues qui peuvent fournir des protocoles antihistaminiques pour supprimer les mastocytes (par exemple, Zyrtec, Zantac, Cromolyn, Singulair). Les avantages d’un test génomique comme le WES qui va au-delà des gènes connus pour causer la dysplasie du tissu conjonctif sont de nouvelles découvertes comme les gènes liés aux muscles maintenant connus pour causer le SDE et la documentation de découvertes fortuites ou secondaires comme les mutations des gènes du cancer du sein et de l’ovaire. La découverte de gènes associés à des symptômes plus graves permet aux patients de connaître les risques pour leur progéniture et de choisir des stratégies de diagnostic génétique prénatal ou préimplantatoire compatibles avec leurs valeurs.
Présentation du cas
Eve est venue voir son médecin à 18 ans, étourdie, faible, anxieuse et malade de l’estomac, si épuisée qu’elle se retirait le vendredi soir pour se reposer tout le week-end. Elle avait été en bonne santé, active en gymnastique et en cheerleading, une fille courageuse et brillante dont la persévérance malgré les blessures valait les applaudissements de ses entraîneurs. Puis vint la grippe dont elle ne s’est jamais remise, des nausées si fortes qu’elle ne pouvait plus manger, une perte de poids, des évanouissements en classe, des réveils nocturnes avec un cœur battant, des réveils matinaux sans sommeil réparateur. Eve a vu de nombreux médecins, même les urgences lorsque les palpitations et la panique lui ont coupé le souffle. Tout allait bien, sauf le stress, disaient-ils, tout était dans sa tête. Calmée mais pas guérie par les pilules, son travail échouant à cause de la fatigue et de la confusion, elle se demandait si elle ne devenait pas folle. Méconnue, sa peau douce et fragile, ses règles dures et abondantes, ses maux de tête quotidiens et, plus frappant encore, une hypermobilité qu’elle et sa famille supposaient normale : « Oui, nous sommes courbés et doublement articulés, n’est-ce pas tout le monde ? ». Eve aurait répondu si on le lui avait demandé.
Heureusement, Eve a cherché un nouveau médecin traitant qui a pu documenter un grand nombre des signes et symptômes énumérés dans le tableau I. Eve a eu des coliques et des reflux précoces avec des difficultés d’allaitement, elle était très souple, appelée le bébé » sans os » par sa mère, et quand elle était plus âgée, elle s’asseyait sur ses genoux avec les jambes étendues derrière elle (la position en » W « ), disant que c’était plus confortable. Au début, elle marchait lentement, tombant fréquemment et paraissant maladroite, mais elle est rapidement devenue active et grimpait partout, faisant grimacer ses parents en mettant un pied autour de son cou ou en faisant le grand écart avec aisance. Mais lorsqu’elle est entrée à l’école primaire, elle a commencé à se plaindre d’arthralgie, ses jambes lui faisant mal la nuit et ses genoux et chevilles la gênant après une activité. Son pédiatre a rassuré ses parents en leur disant qu’elle avait juste des » douleurs de croissance « , mais ils se sont inquiétés davantage lorsqu’elle a essayé de jouer au football et qu’elle n’a cessé de se retourner les chevilles, une entorse ayant nécessité le port d’une botte pendant 2 mois après qu’une fracture de stress ait été constatée.
Eve a dû arrêter le football mais a constaté qu’elle excellait en gymnastique en grandissant, choisissant les exercices au sol en raison d’un mauvais équilibre sur la poutre et d’une tendance à l’arrachement de son épaule sur les barres parallèles. Elle a remarqué que ses articulations sautaient avec le mouvement et les faisait souvent sauter volontairement, car cela semblait soulager la tension et la douleur. Elle s’est inscrite comme pom-pom girl et a réussi ses manœuvres, mais a découvert qu’elle était trop sensible à la chaleur. Les autres filles l’appelaient « tomato face » car elle avait trop chaud pendant les routines, et elle se fatiguait facilement, ayant souvent besoin d’un jour de repos après l’exercice. Ses problèmes digestifs ont refait surface : constipation fréquente et douleurs d’estomac, elle se sentait souvent ballonnée et avait besoin de laxatifs. En sixième année, elle manquait l’école à cause de ses problèmes intestinaux et de ses douleurs articulaires, et se sentait souvent fatiguée et étourdie. Elle ne se sentait pas reposée le matin et faisait souvent des siestes le week-end, ce qui la changeait de son ancien mode de vie actif et enthousiaste. Elle se sentait souvent anxieuse, sans pouvoir l’attribuer à un stress scolaire ou familial particulier, et commençait à avoir des maux de tête le matin qu’elle pensait être dus à son mauvais sommeil. Ses parents l’ont de nouveau emmenée chez le pédiatre et on leur a de nouveau dit qu’il n’y avait rien d’anormal mais le stress scolaire habituel.
Lorsqu’elle avait 14 ans, Eve a eu une mauvaise maladie ressemblant à une grippe et a manqué l’école pendant un mois avec une fatigue sévère, des maux de tête, des nausées, des vomissements et une perte de poids. Elle a eu de terribles vertiges et quelques douleurs à la poitrine, ayant parfois l’impression que son cœur battait la chamade (elle avait des palpitations). Ses parents ont demandé l’avis d’un autre pédiatre, qui a envoyé Eve chez un cardiologue, lequel a pensé qu’elle souffrait de tachycardie en raison du stress et de l’anxiété liés à sa maladie. Il a donné à Eve du propranolol, un bêta-bloquant, pour ralentir son rythme cardiaque, mais elle est devenue encore plus fatiguée et était maintenant alitée une grande partie de la journée. Ses douleurs articulaires se sont intensifiées après sa maladie et elle restait au lit presque toute la journée, se sentant triste et devenant anxieuse lorsque ses parents essayaient de la sortir ou de l’emmener en voiture. Ils l’ont ramenée chez le deuxième pédiatre qui l’a orientée vers un psychiatre et on a commencé à lui administrer du Zoloft pour lutter contre l’anxiété et la dépression. Quelques jours plus tard, Eve a fait une éruption d’urticaire et a dû arrêter ce médicament. Elle a continué à avoir des vertiges et a été examinée par deux autres spécialistes, un gastro-entérologue qui pensait qu’elle pouvait avoir la maladie cœliaque et un oto-rhino-laryngologiste qui a diagnostiqué des vertiges et l’a envoyée en neurologie. Elle a parlé à ce médecin de maux de tête émanant de l’arrière de sa tête et une étude IRM de la tête en décubitus dorsal a été obtenue et s’est avérée normale.
L’appréciation du diagnostic probable de DAA par son médecin traitant a institué une autre série d’orientations vers des sous-spécialités, y compris la neurologie où une étude IRM de la tête en position verticale a démontré la hernie de la déformation de Chiari. Un cardiologue a documenté un test de table basculante anormal ainsi qu’une faible motilité intestinale et a prouvé que son anxiété et sa fatigue due au stress étaient dues à la dysautonomie. Des stratégies d’hydratation nutritionnelle, de sel et de protéines, ainsi qu’un traitement par bêta-bloquant et antihistaminique ont amélioré sa fatigue et ses symptômes intestinaux et diminué ses maux de tête. Il a été décidé que l’hernie de Chiari de 4-5 mm n’était pas suffisante pour nécessiter une intervention chirurgicale, et sa prise en charge psychiatrique a été reconnue comme le traitement des symptômes secondaires d’un trouble médical. Certains de ses sous-spécialistes, sceptiques quant à son diagnostic clinique, ont été convaincus par des tests génétiques qui ont mis en évidence une modification du gène de la chaîne alpha-1 du collagène de type V, abrégé COL5A1, une mutation fréquente chez les patients atteints de SDE. Eve avait hérité de la mutation de sa mère, qui avait eu des symptômes de SDE plus légers et était maintenant capable de mieux les gérer.
Au cours de plusieurs mois, la santé et la vitalité d’Eve ont été rétablies et elle a » retrouvé sa vie « , comme elle et ses parents le diraient. Elle avait encore des douleurs articulaires, de l’anxiété et de la fatigue à l’occasion, mais les » exercices judicieux » pour renforcer la protection musculaire autour de ses articulations, les approches nutritionnelles pour la dysautonomie et les approches de biofeedback pour contrer l’anxiété ont changé sa vie . Tous ces traitements ont été rendus possibles grâce à la reconnaissance de son médecin de famille, qui est maintenant en mesure de coordonner les soins pour Eve et sa mère et de fournir des ressources pour comprendre le SDE et les modifications génétiques sous-jacentes. La transition vers la prochaine génération a été la capacité d’informer Eve de son risque de 50% de transmettre sa mutation COL5A et de certains risques de travail précoce et de saignement post-partum dans toute future grossesse .
- Remvig L, Jensen DV, Ward RC (2007) Épidémiologie de l’hypermobilité articulaire générale et base des critères proposés pour le syndrome d’hypermobilité articulaire bénigne : revue de la littérature. J Rheumatol 34 : 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. In : Le tissu conjonctif et ses désordres héréditaires. B Steinman, PM Royce (Eds) Wiley-Liss : New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby : St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) Le Consortium international sur les syndromes d’Ehlers-Danlos. Am J Med Genet Part C Semin Med Genet 175 : 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomie dans le syndrome d’hypermobilité articulaire. Am J Med 115 : 33-40.
- Pizzo PA1 (2013) Leçons de soulagement de la douleur–une expérience personnelle de troisième cycle. N Engl J Med 369 : 1092-1093.
- Pyeritz RE (2008) Une petite molécule pour une grande maladie. N Engl J Med 358 : 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Chapitre 11 : Séquençage des gènes et du génome : Interpréter la variation génétique au niveau des nucléotides. In : Human Chromosome Variation : Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing–are we there yet ? N Engl J Med 367 : 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Réécriture de l’histoire naturelle de la douleur et des symptômes connexes dans le syndrome d’hypermobilité articulaire/syndrome d’Ehlers-Danlos, type hypermobilité. Am J Med Genet A 12 : 2989-3004.
- Simpson MR (2006) Syndrome d’hypermobilité articulaire bénigne : évaluation, diagnostic et gestion. J Am Osteopath Assoc 106 : 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Plaintes musculo-squelettiques associées à une hypermobilité articulaire généralisée. Ann Rheum Dis 26 : 419-425.
- Wilson GN (2014) Diagnostic génétique présymptomatique et préimplantatoire : Neurologie, NextGenetics, et la prochaine génération. JAMA Neurol 71 : 403-404.
- Castori M, Morlino S, Celletti C (2012) Gestion de la douleur et de la fatigue dans le syndrome d’hypermobilité articulaire (alias syndrome d’Ehlers- Danlos, type hypermobilité) : principes et proposition d’une approche multidisciplinaire. Am J Med Genet A 158 : 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Réécriture de l’histoire naturelle de la douleur et des symptômes associés dans le syndrome d’hypermobilité articulaire/syndrome d’Ehlers-Danlos, type hypermobilité. Am J Med Genet A 161A : 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Traitement multidisciplinaire du handicap dans le syndrome d’Ehlers-Danlos, type hypermobilité/ syndrome d’hypermobilité : une étude pilote utilisant une combinaison de thérapie physique et cognitivo-comportementale sur 12 femmes. Am J Med Genet A 161A : 3005-3011.
- EDS (2018) Fondation nationale du syndrome d’Ehlers-Danlos.
- OMIM (2018) Héritage mendélien en ligne chez l’homme.
- Wilson GN (2014) Analyse de l’exome de la dysplasie du tissu conjonctif : mort et renaissance de la génétique clinique ? Am J Med Genet A 164A : 1209-1212.