Nie powinno nas dziwić, że nawet w częściach genomu, w których nie widzimy oczywiście kodu „funkcjonalnego” (tzn. takiego, który został ewolucyjnie utrwalony w wyniku jakiejś selektywnej przewagi), istnieje pewien rodzaj kodu, ale nie taki jak cokolwiek, co wcześniej uważaliśmy za takie. A co by było, gdyby oprócz jednego wymiaru kodu ATGC robił on coś w trzech wymiarach? Artykuł właśnie opublikowany w BioEssays bada tę tantalizującą możliwość…
Czy to nie wspaniałe mieć naprawdę kłopotliwy problem do zgrzytania, taki, który generuje prawie nieskończoną ilość potencjalnych wyjaśnień. Co powiecie na pytanie „co to całe niekodujące DNA robi w genomach?” – te 98,5% ludzkiego materiału genetycznego, które nie produkuje białek. Prawdę mówiąc, rozszyfrowanie niekodującego DNA czyni wielkie postępy dzięki identyfikacji sekwencji, które są przepisywane na RNA modulujące ekspresję genów, mogą być przekazywane transgeneracyjnie (epigenetyka) lub ustawiają program ekspresji genów komórki macierzystej lub konkretnej komórki tkanki. W wielu genomach znaleziono ogromne ilości powtarzających się sekwencji (pozostałości po starożytnych retrowirusach), i znów, nie kodują one białek, ale przynajmniej istnieją wiarygodne modele tego, co robią w kategoriach ewolucyjnych (od pasożytnictwa genomowego do symbiozy, a nawet „eksploatacji” przez sam genom gospodarza w celu wytworzenia różnorodności genetycznej, na której opiera się ewolucja); Nawiasem mówiąc, niektóre niekodujące DNA tworzy RNA, które wycisza te retrowirusowe sekwencje, i uważa się, że retrowirusowa ingresja do genomów była selektywnym naciskiem na ewolucję interferencji RNA (tzw. RNAi); powtarzające się elementy różnych nazwanych typów i powtórzeń tandemowych obfitują; introny (z których wiele zawiera wyżej wymienione typy niekodujących sekwencji) okazały się kluczowe w ekspresji i regulacji genów, najbardziej uderzająco poprzez alternatywne splicing segmentów kodujących, które oddzielają.
Ciągle jednak jest mnóstwo problemów do rozwiązania, ponieważ chociaż coraz lepiej rozumiemy naturę i pochodzenie znacznej części niekodującego genomu i robimy duże postępy w jego „funkcji” (zdefiniowanej tutaj jako ewolucyjnie wybrany, korzystny wpływ na organizm gospodarza), jesteśmy dalecy od wyjaśnienia tego wszystkiego, a co więcej – patrzymy na to przez obiektyw o bardzo małym powiększeniu, że tak powiem. Jedną z intrygujących rzeczy dotyczących sekwencji DNA jest to, że pojedyncza sekwencja może „kodować” więcej niż jedną informację w zależności od tego, co ją „czyta” i w jakim kierunku – genomy wirusów są klasycznymi przykładami, w których geny odczytywane w jednym kierunku w celu wytworzenia danego białka nakładają się na jeden lub więcej genów odczytywanych w przeciwnym kierunku (tj. z komplementarnej nici DNA) w celu wytworzenia innych białek. Przypomina to trochę tworzenie prostych wiadomości z użyciem słów w odwrotnych parach (tzw. emordnilap). Na przykład: REEDSTOPSFLOW, które przez wyimaginowane urządzenie odczytujące można by podzielić na REED STOPS FLOW. Czytane od tyłu, dałoby to WOLF SPOTS DEER.
Teraz, jeśli jest to ewolucyjnie korzystne dla dwóch wiadomości, aby być zakodowane tak ekonomicznie – jak to ma miejsce w genomach wirusów, które mają tendencję do ewolucji w kierunku minimalnej złożoności pod względem zawartości informacji, zmniejszając w ten sposób niezbędne zasoby do reprodukcji – wtedy wiadomości same ewoluują z wysokim stopniem ograniczenia. Co to oznacza? Cóż, moglibyśmy sformułować nasz oryginalny przykładowy komunikat jako RUSH-STEM IMPEDES CURRENT, który zawierałby tę samą istotną informację co REED STOPS FLOW. Jednak ta wiadomość, czytana w odwrotnym kierunku (lub nawet w tym samym sensie, ale w różnych fragmentach) nie koduje niczego dodatkowego, co byłoby szczególnie znaczące. Prawdopodobnie jedynym sposobem jednoczesnego przekazania obu informacji zawartych w oryginalnych wiadomościach jest samo sformułowanie REEDSTOPSFLOW: to jest bardzo ograniczony system! Rzeczywiście, gdybyśmy przestudiowali wystarczająco dużo przykładów zwrotów odwróconej pary w języku angielskim, zobaczylibyśmy, że są one, na ogół, złożone z raczej krótkich słów, a w sekwencjach brakuje pewnych jednostek języka, takich jak artykuły (the, a); gdybyśmy przyjrzeli się bliżej, moglibyśmy nawet wykryć większą niż przeciętnie reprezentację pewnych liter alfabetu w takich wiadomościach. Widzielibyśmy to jako odchylenia w użyciu słów i liter, które a priori pozwoliłyby nam na identyfikację takich „dwufunkcyjnych” fragmentów informacji.
Powróćmy teraz do „liter”, „słów” i „informacji” zakodowanych w genomach. Dla dwóch odrębnych kawałków informacji, które mają być zakodowane w tym samym kawałku sekwencji genetycznej, podobnie, oczekiwalibyśmy, że ograniczenia przejawiają się w tendencyjności użycia słów i liter – analogie, odpowiednio, do sekwencji aminokwasów tworzących białka i ich trzyliterowego kodu. Stąd sekwencja DNA może kodować białko i, dodatkowo, coś jeszcze. To „coś jeszcze”, zdaniem Giorgio Bernardiego, to informacja, która kieruje upakowaniem ogromnej długości DNA w komórce do stosunkowo maleńkiego jądra. Przede wszystkim jest to kod, który kieruje wiązaniem białek opakowujących DNA, znanych jako histony. Bernardi odnosi się do tego jako „kod genomowy” – kod strukturalny, który określa kształt i zagęszczenie DNA w wysoce zagęszczonej formie znanej jako „chromatyna”.
Ale nie zaczęliśmy od wyjaśnienia dla niekodującego DNA, a nie sekwencji kodujących białka? Tak, a w długich odcinkach niekodującego DNA widzimy informacje wykraczające poza zwykłe powtórzenia, powtórzenia tandemowe i pozostałości starożytnych retrowirusów: istnieje rodzaj kodu na poziomie preferencji dla pary GC chemicznych zasad DNA w porównaniu z AT. Jak opisuje Bernardi, syntetyzując swoją i innych przełomową pracę, w podstawowych sekwencjach genomu eukariotycznego, zawartość GC w strukturalnych jednostkach organizacyjnych genomu zwanych „izochorami” wzrosła podczas ewolucyjnego przejścia między tak zwanymi organizmami zimnokrwistymi i ciepłokrwistymi. I, co fascynujące, ta tendencyjność sekwencji pokrywa się z sekwencjami, które są znacznie bardziej ograniczone w funkcji: są to właśnie sekwencje kodujące białka, o których była mowa wcześniej, i to one – bardziej niż interweniujące sekwencje niekodujące – są wskazówką do „kodu genomowego”.
Sekwencje kodujące białka są również upakowane i zagęszczone w jądrze – szczególnie, gdy nie są „w użyciu” (tj, są transkrybowane, a następnie tłumaczone na białko) – ale zawierają również względnie stałą informację o dokładnej tożsamości aminokwasów, w przeciwnym razie nie udałoby im się prawidłowo kodować białek: ewolucja działałaby na takie mutacje w wysoce negatywny sposób, czyniąc je niezwykle mało prawdopodobnymi do przetrwania i bycia widocznymi dla nas. Ale kod aminokwasów w DNA ma mały „haczyk”, który wyewoluował w najprostszych organizmach jednokomórkowych (bakterie i archaidy) miliardy lat temu: kod jest częściowo redundantny. Na przykład, aminokwas treonina może być zakodowany w eukariotycznym DNA na nie mniej niż cztery sposoby: ACT, ACC, ACA lub ACG. Trzecia litera jest zmienna, a więc „dostępna” do zakodowania dodatkowej informacji. Tak właśnie dzieje się w przypadku „kodu genomowego”, który w tym przypadku u organizmów ciepłokrwistych preferuje formy ACC i ACG. Stąd, wysokie ograniczenie tego dodatkowego „kodu” – który jest również widoczny w częściach genomu, które nie podlegają takiemu ograniczeniu jak sekwencje kodujące białka – jest narzucone przez upakowanie sekwencji kodujących białka, które ucieleśniają dwa zestawy informacji jednocześnie. Jest to analogiczne do naszego przykładu silnie ograniczonej podwójnej sekwencji informacyjnej REEDSTOPSFLOW.
Ważne jest jednak, że ograniczenie nie jest tak ścisłe jak w naszym przykładzie z języka angielskiego z powodu nadmiarowości trzeciej pozycji tripletu kodu aminokwasów: lepszą analogią byłoby SHE*ATE*STU*, gdzie gwiazdka oznacza zmienną literę, która nie robi żadnej różnicy maszynie czytającej trzyliterowy składnik czteroliterowej wiadomości. Można sobie wyobrazić drugi poziom informacji utworzony przez dodanie „D” w tych punktach z gwiazdką, aby utworzyć SHEDATEDSTUD (SHE DATED STUD). Następnie wyobraźmy sobie drugą maszynę czytającą, która szuka znaczących fraz o „wrażliwej naturze”, zawierających ponadprzeciętną koncentrację Ds. Ta maszyna czytająca ma ze sobą maszynę składającą, która umieszcza coś w rodzaju kołka przy każdym D, zaginając wiadomość o 120 stopni w pewnej płaszczyźnie. punkt, w którym wiadomość powinna być zagięta o 120 stopni w tej samej płaszczyźnie, otrzymamy bardziej zwartą, trójkątną wersję. W genomach eukariotycznych tendencyjność sekwencji GC, która ma odpowiadać za kondensację strukturalną, rozciąga się na sekwencje niekodujące, z których część ma zidentyfikowaną aktywność, choć są one mniej ograniczone sekwencyjnie niż DNA kodujące białka. Tam kieruje ich kondensacją poprzez nukleosomy zawierające histony, tworząc chromatynę.
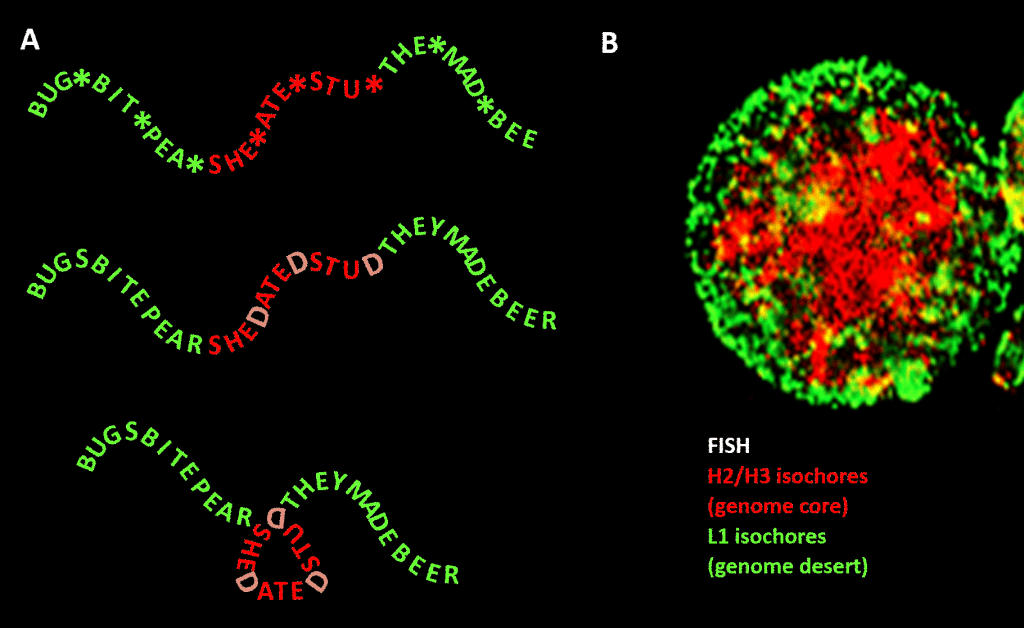
Rysunek. Analogia między kondensacją wiadomości opartej na słowach a kondensacją genomowego DNA w jądrze komórkowym. Panel A: Informacja w informacji, ciąg słów ze zmienną czwartą spacją, która po wypełnieniu poszczególnymi literami generuje dalszą wiadomość. Jedna wiadomość jest odczytywana przez maszynę odczytującą trzy litery; druga przez maszynę odczytującą, która może interpretować informacje rozciągające się do czwartej zmiennej pozycji sekwencji. Drugi czytnik rozpoznaje „wrażliwą” informację, która powinna być ukryta, i w miejscach, gdzie na czwartej pozycji pojawia się „D”, składa ciąg słów, ściskając w ten sposób „wrażliwą” część i usuwając ją z pola widzenia. Jest to analogia do zasady genomowej kompresji 3D przez chromatynę, przedstawionej w panelu B: obraz fluorescencyjny (poprzez Fluorescence In-Situ Hybridization – FISH) jądra komórkowego. Izochory H2/H3, których zawartość GC wzrosła podczas ewolucji od zimnokrwistych do ciepłokrwistych kręgowców, są ściśnięte w chromatynowym rdzeniu, pozostawiając izochory L1 (z niższą zawartością GC) na peryferiach w mniej skondensowanym stanie. Kod genomowy” zawarty w trakcjach genomu o wysokiej zawartości GC jest, zdaniem Bernardiego, odczytywany przez maszynerię pozycjonowania nukleosomów w komórce i interpretowany jako sekwencja, która ma być silnie skompresowana w euchromatynie. Podziękowania: Panel A: koncepcja i produkcja figur: Andrew Moore; Panel B: Wzór FISH izochor H2/H3 i L1 z limfocytu indukowanego przez PHA – dzięki uprzejmości S. Saccone – jak odtworzono w Ref. .]
Te regiony DNA mogą być wtedy traktowane jako strukturalnie ważne elementy w tworzeniu prawidłowego kształtu i separacji skondensowanych sekwencji kodujących w genomie, niezależnie od jakiejkolwiek innej możliwej funkcji, jaką mają te niekodujące sekwencje: w istocie byłoby to „wyjaśnienie” dla utrzymywania się w genomach sekwencji, którym nie można przypisać żadnej „funkcji” (w sensie ewolucyjnie wybranej aktywności) (lub, przynajmniej, żadnej istotnej funkcji).
Ostatnia analogia – tym razem znacznie bliżej spokrewniona – może być bardzo sekwencje aminokwasów w dużych białkach, które robią różne skręty, obroty, fałdy itp. Możemy podziwiać tak skomplikowane struktury i zapytać „ale czy muszą być one dość skomplikowane dla ich funkcji?”. Cóż, może muszą, aby skondensować i umieścić części białka w dokładnej orientacji i miejscu, które generuje trójwymiarową strukturę, która została pomyślnie wybrana przez ewolucję. Ale z wiedzą, że „kod genomowy” nakłada się na sekwencje kodujące białka, możemy nawet zacząć podejrzewać, że jest inny selektywny nacisk w pracy, jak również…
Andrew Moore, Ph.D.
Editor-in-Chief, BioEssays