Es sollte uns nicht überraschen, dass selbst in Teilen des Genoms, in denen wir offensichtlich keinen „funktionalen“ Code sehen (d.h. einen, der evolutionär als Ergebnis eines Selektionsvorteils festgelegt wurde), eine Art von Code vorhanden ist, aber nicht im Sinne von etwas, das wir bisher als solchen betrachtet haben. Und was wäre, wenn er neben der einen Dimension des ATGC-Codes auch etwas in drei Dimensionen tun würde? Ein gerade in BioEssays veröffentlichter Artikel erforscht diese verlockende Möglichkeit…
Ist es nicht wunderbar, ein wirklich verwirrendes Problem zu haben, an dem man knabbern kann, eines, das fast endlose mögliche Erklärungen hervorbringt. Wie wäre es mit der Frage: „Was macht all die nicht codierende DNA in den Genomen?“ – diese 98,5 % des menschlichen Erbguts, aus denen keine Proteine entstehen. Fairerweise muss man sagen, dass die Entschlüsselung der nichtcodierenden DNA große Fortschritte macht, da Sequenzen identifiziert wurden, die in RNAs umgeschrieben werden, die die Genexpression modulieren, transgenerational weitergegeben werden können (Epigenetik) oder das Genexpressionsprogramm einer Stammzelle oder einer bestimmten Gewebezelle festlegen. In vielen Genomen wurden riesige Mengen an Wiederholungssequenzen (Überbleibsel alter Retroviren) gefunden, die wiederum nicht für Proteine kodieren, aber zumindest gibt es glaubwürdige Modelle dafür, was sie in evolutionärer Hinsicht tun (von genomischem Parasitismus über Symbiose bis hin zur „Ausbeutung“ durch das Wirtsgenom selbst, um die genetische Vielfalt zu erzeugen, auf der die Evolution beruht); Nebenbei bemerkt, stellt einige nicht-kodierende DNA RNAs her, die diese retroviralen Sequenzen zum Schweigen bringen, und es wird angenommen, dass das Eindringen von Retroviren in Genome der Selektionsdruck für die Evolution der RNA-Interferenz (so genannte RNAi) war; repetitive Elemente verschiedener Typen und Tandem-Wiederholungen gibt es zuhauf; Introns (von denen viele die oben erwähnten Arten von nicht-kodierenden Sequenzen enthalten) haben sich als entscheidend für die Genexpression und -regulation erwiesen, am auffälligsten durch alternatives Spleißen der kodierenden Segmente, die sie trennen.
Noch immer gibt es eine Menge Probleme, an denen wir zu knabbern haben, denn obwohl wir die Natur und den Ursprung eines Großteils des nichtcodierenden Genoms immer besser verstehen und große Fortschritte bei der Erforschung seiner „Funktion“ (hier definiert als evolutionär ausgewählte, vorteilhafte Wirkung auf den Wirtsorganismus) machen, sind wir weit davon entfernt, alles zu erklären, und – was noch wichtiger ist – wir betrachten es sozusagen mit einer Linse mit sehr geringer Vergrößerung. Eines der faszinierenden Dinge an DNA-Sequenzen ist, dass eine einzelne Sequenz mehr als eine Information „kodieren“ kann, je nachdem, was sie „liest“ und in welche Richtung – virale Genome sind klassische Beispiele, bei denen sich Gene, die in eine Richtung gelesen werden, um ein bestimmtes Protein zu produzieren, mit einem oder mehreren Genen überschneiden, die in die entgegengesetzte Richtung gelesen werden (d. h. vom komplementären DNA-Strang), um verschiedene Proteine zu produzieren. Das ist ein bisschen so, wie wenn man einfache Nachrichten mit umgekehrten Wortpaaren schreibt (ein so genannter Emordnilap). Zum Beispiel: REEDSTOPSFLOW, das durch ein imaginäres Lesegerät in REED STOPS FLOW zerlegt werden könnte. Rückwärts gelesen ergäbe das WOLF SPOTS DEER.
Wenn es nun von evolutionärem Vorteil ist, dass zwei Botschaften so sparsam kodiert sind – wie es bei viralen Genomen der Fall ist, die dazu neigen, sich in Richtung minimaler Komplexität in Bezug auf den Informationsgehalt zu entwickeln und damit die notwendigen Ressourcen für die Reproduktion zu reduzieren – dann entwickeln sich die Botschaften selbst mit einem hohen Grad an Zwang. Was ist damit gemeint? Nun, wir könnten unsere ursprüngliche Beispielnachricht als RUSH-STEM IMPEDES CURRENT formulieren, was dieselbe wesentliche Information enthalten würde wie REED STOPS FLOW. Allerdings enthält diese Nachricht, wenn sie umgekehrt gelesen wird (oder sogar im gleichen Sinne, aber in anderen Abschnitten), keine zusätzlichen Informationen, die besonders bedeutsam sind. Wahrscheinlich ist die einzige Möglichkeit, die beiden Informationen in den Originalnachrichten gleichzeitig zu übermitteln, der Wortlaut REEDSTOPSFLOW: Das ist ein sehr eingeschränktes System! Wenn wir genügend Beispiele für umgekehrte Satzpaare im Englischen untersuchen würden, würden wir feststellen, dass sie im Großen und Ganzen aus eher kurzen Wörtern bestehen und dass in den Sequenzen bestimmte sprachliche Einheiten wie Artikel (the, a) fehlen; wenn wir genauer hinsehen würden, könnten wir sogar feststellen, dass bestimmte Buchstaben des Alphabets in solchen Nachrichten überdurchschnittlich häufig vorkommen. Wir würden dies als Verzerrungen im Wort- und Buchstabengebrauch ansehen, die es uns a priori erlauben würden, solche „Doppelfunktions“-Informationen zu identifizieren.
Wenden wir uns nun wieder den „Buchstaben“, „Wörtern“ und „Informationen“ zu, die in Genomen kodiert sind. Wenn zwei unterschiedliche Informationen in ein und demselben Stück genetischer Sequenz kodiert werden, würden wir erwarten, dass sich die Zwänge in einer verzerrten Verwendung von Wörtern und Buchstaben manifestieren – Analogien für Aminosäuresequenzen, die Proteine bilden, und deren Drei-Buchstaben-Code. Eine DNA-Sequenz kann also für ein Protein und darüber hinaus für etwas anderes kodieren. Dieses „Etwas“ ist nach Giorgio Bernardi die Information, die das Verpacken der enormen Länge der DNA in einer Zelle in den relativ winzigen Zellkern steuert. In erster Linie handelt es sich dabei um den Code, der die Bindung der als Histone bekannten DNA-Verpackungsproteine steuert. Bernardi nennt dies den „genomischen Code“ – einen strukturellen Code, der die Form und die Verdichtung der DNA in die hochverdichtete Form, die als „Chromatin“ bekannt ist, definiert.
Aber haben wir nicht mit einer Erklärung für nicht-kodierende DNA begonnen, nicht für Protein-kodierende Sequenzen? Ja, und in den langen Abschnitten der nicht-kodierenden DNA sehen wir Informationen, die über bloße Wiederholungen, Tandem-Wiederholungen und Überreste alter Retroviren hinausgehen: Es gibt eine Art von Code auf der Ebene der Bevorzugung des GC-Paares chemischer DNA-Basen gegenüber AT. Wie Bernardi in einer Zusammenfassung seiner und anderer bahnbrechender Arbeiten darlegt, nahm der GC-Gehalt in den Kernsequenzen des eukaryotischen Genoms in strukturellen Organisationseinheiten des Genoms, den so genannten „Isochoren“, während des evolutionären Übergangs zwischen so genannten kaltblütigen und warmblütigen Organismen zu. Faszinierenderweise überschneidet sich dieser Sequenz-Bias mit Sequenzen, die in ihrer Funktion sehr viel eingeschränkter sind: Dies sind die bereits erwähnten proteinkodierenden Sequenzen, die – mehr als die dazwischen liegenden nicht-kodierenden Sequenzen – den Hinweis auf den „genomischen Code“ darstellen.
Proteinkodierende Sequenzen sind auch im Zellkern gepackt und verdichtet – insbesondere wenn sie nicht „in Gebrauch“ sind (d.h., Sie enthalten aber auch relativ konstante Informationen über die genauen Aminosäure-Identitäten, da sie sonst nicht in der Lage wären, Proteine korrekt zu kodieren: Die Evolution würde sich auf solche Mutationen äußerst negativ auswirken, so dass es extrem unwahrscheinlich wäre, dass sie fortbestehen und für uns sichtbar sind. Der Aminosäurencode in der DNA hat jedoch einen kleinen „Haken“, der sich bei den einfachsten einzelligen Organismen (Bakterien und Archaeen) vor Milliarden von Jahren entwickelt hat: Der Code ist teilweise redundant. Die Aminosäure Threonin zum Beispiel kann in der eukaryontischen DNA auf nicht weniger als vier Arten kodiert werden: ACT, ACC, ACA oder ACG. Der dritte Buchstabe ist variabel und somit „verfügbar“ für die Codierung zusätzlicher Informationen. Genau dies geschieht bei der Erstellung des „genomischen Codes“, wobei in diesem Fall die Formen ACC und ACG bei warmblütigen Organismen bevorzugt werden. Die starke Einschränkung dieses zusätzlichen „Codes“ – die auch in Teilen des Genoms zu beobachten ist, die als proteinkodierende Sequenzen nicht unter einer solchen Einschränkung stehen – wird also durch die Verpackung proteinkodierender Sequenzen verursacht, die zwei Informationsgruppen gleichzeitig verkörpern. Dies ist analog zu unserem Beispiel der hochgradig eingeschränkten Doppelinformationssequenz REEDSTOPSFLOW.
Wichtig ist jedoch, dass die Einschränkung nicht so streng ist wie in unserem englischsprachigen Beispiel wegen der Redundanz der dritten Position des Triplett-Codes für Aminosäuren: eine bessere Analogie wäre SHE*ATE*STU*, wobei das Sternchen für einen variablen Buchstaben steht, der für die Maschine, die die Drei-Buchstaben-Komponente der Vier-Buchstaben-Nachricht liest, keinen Unterschied macht. Man könnte sich dann eine zweite Informationsebene vorstellen, die durch Hinzufügen eines „D“ an diesen Sternchenpunkten gebildet wird, so dass SHEDATEDSTUD (SHE DATED STUD) entsteht. Als Nächstes stellen Sie sich eine zweite Lesemaschine vor, die nach bedeutungsvollen Sätzen „empfindlicher Natur“ sucht, die eine überdurchschnittliche Konzentration von Ds enthalten. Diese Lesemaschine führt eine Faltmaschine mit sich, die an jedem D eine Art Stift anbringt, der die Nachricht um 120 Grad in einer Ebene knickt. einen Punkt, an dem die Nachricht um 120 Grad in derselben Ebene gebogen werden sollte, würden wir am Ende eine kompaktere, dreieckige Version erhalten. In eukaryotischen Genomen erstreckt sich der GC-Sequenz-Bias, der für die strukturelle Kondensation verantwortlich sein soll, auch auf nicht-kodierende Sequenzen, von denen einige identifizierte Aktivitäten aufweisen, obwohl sie in ihrer Sequenz weniger eingeschränkt sind als proteinkodierende DNA. Dort steuert sie deren Kondensation über histonhaltige Nukleosomen zur Bildung von Chromatin.
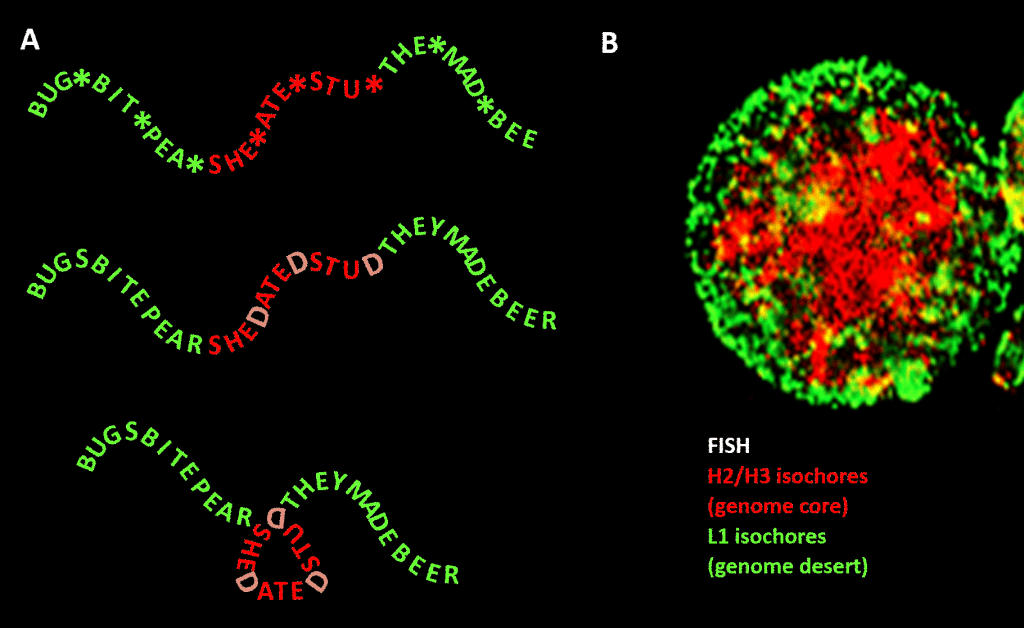
Abbildung. Analogie zwischen der Kondensation einer wortbasierten Nachricht und der Kondensation der genomischen DNA im Zellkern. Tafel A: Information in der Information, eine Folge von Wörtern mit einem variablen vierten Leerzeichen, das, wenn es mit bestimmten Buchstaben gefüllt wird, eine weitere Nachricht erzeugt. Eine Nachricht wird von einer Lesemaschine mit drei Buchstaben gelesen, die andere von einer Lesemaschine, die Informationen interpretieren kann, die bis zur vierten variablen Stelle der Sequenz reichen. Die zweite Lesemaschine erkennt „sensible“ Informationen, die verborgen werden sollten, und faltet an den Stellen, an denen ein „D“ an der vierten Stelle auftaucht, die Wortfolge, wodurch der „sensible“ Teil komprimiert wird und nicht mehr sichtbar ist. Dies ist eine Analogie für das Prinzip der genomischen 3D-Kompression durch Chromatin, wie es in Tafel B dargestellt ist: ein Fluoreszenzbild (mittels Fluoreszenz-In-Situ-Hybridisierung – FISH) des Zellkerns. Die H2/H3-Isochoren, deren GC-Gehalt während der Evolution von kaltblütigen zu warmblütigen Wirbeltieren zunahm, werden zu einem Chromatinkern komprimiert, während die L1-Isochoren (mit geringerem GC-Gehalt) an der Peripherie in einem weniger komprimierten Zustand verbleiben. Der „genomische Code“, der in den GC-reichen Abschnitten des Genoms verkörpert ist, wird nach Bernardi von der Nukleosomen-Positionierungsmaschinerie der Zelle gelesen und als Sequenz interpretiert, die im Euchromatin stark komprimiert werden muss. Danksagung: Tafel A: Konzept und Erstellung der Abbildungen: Andrew Moore; Tafel B: Ein FISH-Muster von H2/H3- und L1-Isochoren aus einem durch PHA induzierten Lymphozyten – mit freundlicher Genehmigung von S. Saccone – wie in Ref. wiedergegeben]
Diese DNA-Regionen können dann als strukturell wichtige Elemente bei der Bildung der korrekten Form und Trennung kondensierter kodierender Sequenzen im Genom betrachtet werden, unabhängig von jeder anderen möglichen Funktion, die diese nicht-kodierenden Sequenzen haben: Im Wesentlichen wäre dies eine „Erklärung“ für das Fortbestehen von Sequenzen in Genomen, denen keine „Funktion“ (im Sinne einer evolutionär ausgewählten Aktivität) zugeschrieben werden kann (oder zumindest keine wesentliche Funktion).
Eine letzte Analogie – diesmal sehr viel enger verwandt – könnten die Aminosäuresequenzen in großen Proteinen sein, die eine Vielzahl von Drehungen, Wendungen, Faltungen usw. ausführen. Wir mögen über solch komplizierte Strukturen staunen und uns fragen: „Aber müssen sie für ihre Funktion wirklich so kompliziert sein?“ Nun, vielleicht müssen sie das, um Teile des Proteins in der exakten Ausrichtung und an der exakten Stelle zu kondensieren und zu positionieren, die die dreidimensionale Struktur erzeugt, die von der Evolution erfolgreich ausgewählt wurde. Aber mit dem Wissen, dass sich der „genomische Code“ mit den proteinkodierenden Sequenzen überschneidet, könnten wir sogar den Verdacht hegen, dass noch ein anderer Selektionsdruck am Werk ist…
Andrew Moore, Ph.D.
Chefredakteur, BioEssays