Nyckelord
hypermobilitet, bindvävsdysplasi, ehlers-danlos syndrom, beighton-skalan, dysautonomi, posturalt ortostatiskt takykardisyndrom, irritable bowel syndrome, mastcellsaktiveringsstörning, articulo-autonomisk dysplasi
Introduktion
De 20 procent av kvinnorna och 10 procent av männen vars vävnader är mer flexibla och bräckliga än genomsnittet är kraftigt underkända och tragiskt felbehandlade. Få möjligheter till förebyggande och behandling är större än att känna igen hypermobilitet, som lätt kan upptäckas genom att be patienten trycka tummen mot underarmen (figur 1). Detta tecken kan visa vägen till en mer detaljerad utvärdering som ligger väl inom allmänläkarens område, eftersom deras breda kliniska kunskap stämmer väl överens med de multisystemproblem som kan uppstå till följd av slapp bindväv. Här går jag igenom karaktären hos hypermobilitetsstörningar, visar att allmän snarare än subspecialiserad kunskap kan leda till livsavgörande terapier för dessa patienter och visar hur genetisk testning för in symptomatiska beskrivningar som fibromyalgi eller kroniskt trötthetssyndrom i precisionsmedicinens urskiljande ljus.
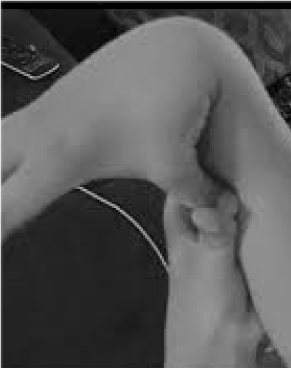
Figur 1. Hypermobilitet.
Flera hinder måste övervinnas innan hypermobilitetspatienter kan dra nytta av fördelarna med allmänmedicin. Det första härrör innebär att dermatologerna Ehlers och Danlos banade väg för ett banbrytande men kortsiktigt fokus genom att prägla otroliga och exotiska hudutvidgningar som den missvisande symbolen för bindvävslaxitetssjukdomar. Ett andra hinder är den ökande klyftan mellan praktisk och genetisk medicin, där den förstnämnda lämpligen koncentrerar sig på vanliga sjukdomar medan den sistnämnda tills nyligen använde specialiserad teknik för att beskriva de sällsynta. De obskyra eponymerna med skrämmande subtyper och tekniska termer är som små provinser i ett oinbjudande främmande land som ingen global resenär tar sig tid att kartlägga. För att motverka dessa hinder behövs ett praktiskt tillvägagångssätt för hypermobilitetssjukdomar som är inriktat på att känna igen sig genom anamnes och fysisk undersökning, och som överlåter teknikaliteterna kring eponymer, subtyper och genetisk testning till subspecialister som kontaktas efter det viktiga första steget, dvs. den förmodade diagnosen. När den flexibla patienten väl är identifierad och hänvisad förblir han eller hon beroende av sin primärläkare, för det är bara han eller hon som kan integrera rekommendationer och testresultat i det allmänna medicinska hemmet.
Terminologi
En ökad rörelseomfång vid flera artikulations- (led-) ställen förekommer vid ett stort antal sjukdomar, allt från kromosomrubbningar som Downs syndrom till sjukdomar som Ehlers-Danlos syndrom (EDS) som är kända för sin hypermobilitet. McKusick populariserade denna kategori av sjukdomar som är kända för vävnadslöshet genom att gruppera dem som ärftliga sjukdomar i bindväven, inklusive Marfans syndrom som han fokuserade på som kardiolog. År 1977 organiserade dr Peter Beighton, känd för sin skala för att mäta flexibilitet, den mellanliggande litteraturen genom att postulera 6-7 EDS-typer, som nu förenklas av nya riktlinjer som EDS klassiska, vaskulära och hypermobila . Det står nu klart att dessa typer har betydande överlappning av symtom mellan varandra och relaterade sjukdomar som osteogenesis imperfect (med många frakturer), Marfans syndrom (utvidgning/dissektion av stora kärl), Sticklers syndrom (ögon- och artritförändringar) och många andra.
Snarare än att ägna sig åt särskilda sjukdomar finner jag det nyttigare att fokusera på två stora sjukdomsprocesser från bindvävsdysplasier, illustrerade av de många symtom som anges i tabell 1. Den första är den slitna ledsmärtan (osteoartrit) som skiljer sig från de svullna, röda lederna vid inflammatorisk (autoimmun eller inflammatorisk) artrit som förekommer vid reumatoid artrit, lupus, Sjögrens syndrom eller ankyloserande spondylit. Den andra är den obalans i det autonoma nervsystemet (dysautonomi) som orsakas av elastiska blodkärl vid hypermobilitetssjukdomar . Distensibla kärl orsakar blodsamlingar i bäcken och ben, med lägre blodtryck och yrsel/svaghet när man står upp (ortostatisk hypotension). Med tiden aktiverar det minskade blodflödet till huvudet den sympatiska armen i det autonoma nervsystemet (adrenalinarm ”fly eller slåss”), vilket leder till flera problem: 1) posturalt ortostatiskt takykardisyndrom (POTS) med takykardi, ångest, kronisk trötthet och ”hjärndimma” (intervaller med nedsatt fokus och minne), 2) irritabelt tarmsyndrom på grund av hämning av tarmmotiliteten (parasympatiskt undertryckande) med förstoppning omväxlande med diarré, uppblåsthet, reflux, magsmärta, dysfagi, illamående, och 3) mastcellsaktiveringsstörning med övergående utslag, nässelfeber/reaktiv hud, astma/ andfåddhet och intolerans mot mat/läkemedel. Jag har utarbetat en bredare term som underlättar erkännandet av alla hypermobilitetsstörningar och som erkänner de två huvudsakliga konsekvenserna av led-/vävnadslöshet: articulo-autonomisk dysplasi eller ledtrötthetskomplex.
Tabell 1. Typiska fynd och deras frekvens vid hypermobilitetssyndrom. *Från 320 kvinnliga och 54 manliga patienter över 12 år som bedömdes med detaljerade frågeformulär 2017; TMJ, temporomandibulär led; POTS, posturalt ortostatiskt takykardisyndrom. Symptomlista delvis anpassad från Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
HISTORIA (procentsatser)*
Som barn: Kolik (28), matningssvårigheter (19,4); klumpig, många fall (29,1)
Barn/tonåring: Långsam viktökning (12); medveten om smidighet (55,6); visade upp ledtricks (53,6)
tidig ledvärk (42,1); tonårsaktiviteter begränsade på grund av smärta-skada (55,4); tidiga glasögon (46,8)
Ortodonti (61,4 %) tandköttsbesvär (11,3); dålig emalj/många hål (31,6)
Ledproblem: Ledsen viktökning (12); medveten om smidighet (55,6); visade upp ledtricks (53,6); många hål (31,6): Smärta i minst två leder (80,3); ledoperationer (33); många stukningar (26); frakturer >2 (31,5)
ryggradsdiskbråck (29,3)
Skelettproblem: Skolios (31); pectus (2,2); gång – tångtående in/ut (9,3); platta fötter (15,5)
Hud: Lätt att få blåmärken (61); stretchig hud (15); sammetslen (18); ovanliga ärr (36,3)
striae före graviditet (73); långsam läkning (32,4)
Genitourinary (kvinnor): Menorragi (61,2); endometrios (11,3); ovariecystor (55)
polycystiskt ovariesyndrom (8,5); problem med urinblåsan (46,1); bråck i ljumsken/femoralen (11,5)
Neuromuskulärt: Migrän (51,7); daglig huvudvärk (62,3); behövde medicin mot huvudvärk (45)
Chiari-deformation (10,6); neurokirurgi (2,9); domningar/prickningar (48,7); dålig balans (48,6)
neuropatisk skjutande smärta (37,3); muskelsmärta/spasmer (51,7); muskelsvaghet (32,4)
GI/bälgar: Förstoppning-diarré (67,1); uppblåsthet-reflux-magsmärta (57,3)
Problem med gallblåsan (12,9); svårt att svälja (23); frekvent illamående (44,9)
Hjärta: Hjärtat: Mitralisklaffprolaps (8,7); aneurysm (0,9); annan arytmi än takykardi (6,8)
POTS: Svårt att stå upp (69,8); synkope (34,5); kronisk trötthet (81,8); sömnsvårigheter (43,5)
hjärndimma (dåligt minne eller fokus ibland-70,8); värme-/kylakänslighet (77,5); onormal svettning (25); takykardi (72,8); ångest- och panikattacker (61,3); saltlust (51,2)
Immun/mastcell: Övergående utslag (33,4 %); nässelfeber/reaktiv hud (45,9 %); astma/andnöd (39,3); intolerans mot mat och läkemedel (61,5 %)
Labb/Bildtagning:: Läkemedel: (1,5 %); (1,5 %); (1,5 %); (1,5 %); (1,5 %): Låg bentäthet (2,1), lågt D-vitamin (28,7), lågt B12-vitamin (2,7), hypotyreos (11,5), lågt ferritin (1,7), lågt järn (6,7), förhöjda antikärnantikroppar (7,7).1)
Genomsnittligt antal fynd av 80: kvinnor >12 (35,4 fynd) män >12 (21,5 fynd)
Fysiskt (procent)
Bygg: Lång höjd >90:e percentilen (55,2); tungt-BMI >28 (21,3); smalt-BMI <19 (14,2)
Marfanoid habitus (44); arachnodactyly (46,4); Walker-Murdoch tecken närvarande (40,2)
Ansikte: Lång (25,7); stram nedre del av ansiktet (5,4); blågrå sclera (1,6); hög gom (53,5)
Hud: Mjuk (81,4); genomskinlig (23,8); elastiskt lyft >1 tum stort veck runt käken på mitten av underarmen (51,3)
Vecken består av det yttre (epidermala) lagret (35.4); ovanliga ärr (42,1 %)
Beighton-poäng: 0-3 poäng (8,4); 4-6 poäng (32,6); 7-8 poäng (38,8); 9 av 9 poäng (20,2)
Andra manövrer: Andra manövrer: Samla händerna över axeln bakom ryggen (68)
uppåtriktat bönetecken bakom ryggen (71,3); föra handen runt ryggen för att röra vid naveln (19,5)
Skelettet: Nackkyfos (41,2); skolios (22,3); lordos (32,8); platta fötter (39,6)
Gångstil – att gå inåt eller utåt (28,3)
Neurologiskt: Muskelsvaghet (11,2); minskad muskelmassa (8,8)
dålig balans vid tandemgång (33,4)
Genomsnittligt antal fynd av 40: kvinnor >12 (17,9 fynd) män >12 (15.7 fynd)
Artikulo-autonomisk dysplasi (AAD)
En term som ”artikulo-autonomisk dysplasi” eller AAD kan förenhetliga tillvägagångssättet när det gäller bindvävslaxitet/hypermobilitetsstörningar och motverka tre problem som komplicerar igenkännandet av dem: 1) Många läkare och genetiker betraktar bindvävsdysplasier som sällsynta och extrema sjukdomar när de i själva verket drabbar cirka 1 % av befolkningen. 2) Riktlinjerna för diagnostik av EDS, som visserligen förenklar de viktigaste EDS-typerna som klassisk, vaskulär och hypermobilitet, skapar fortfarande förvirring genom att de hänvisar till många mindre typer som bildar ett överlappande symtomspektrum. 3) Den nästan obligatoriska dysautonomi som åtföljer EDS och besläktade sjukdomar (tabell 1) utelämnas som ett diagnostiskt kriterium, vilket är som att beskriva en elefant utan att nämna dess bål. AAD betecknar det oundvikliga fysiologiska sambandet mellan lösdrift och dysautonomi på samma sätt som de metabola eller hepatiska syndromen beskriver hur fetma leder till leversjukdom/diabetes eller hur leversvikt orsakar njursjukdom.
Som de flesta medicinska mönster eller syndrom, inklusive EDS, är AAD ett symtomkomplex med många underliggande orsaker. ”Articulo” hänvisar till leden, med erkännande av att en sjukdom i någon ledkomponent – hud, nerv, muskel, ben, bindväv, blodkärl – orsakar ett liknande mönster av ledvävnadslöshet med autonom obalans. Det kommer att finnas hudbristfällighet, minskad bentäthet med benägenhet för frakturer, muskelsvaghet med smärta (myalgi), neuropati med domningar/prickningar, karpaltunnel eller regionala smärtsyndrom, autonoma förändringar, inklusive takykardi, ångest, icke-restorativ sömn, tarminfektion, nässelutslag/utslag, intolerans mot mat och läkemedel, som alla förekommer i varierande omfattning även inom specifika ”typer” av EDS. Jag har inte funnit några stora skillnader mellan patienter med särskilda typer av EDS, vilket illustreras av mer frekventa hudförändringar i typ I klassisk cEDS jämfört med större ledflexibilitet i typ III hypermobil hEDS. Frekvenserna av symtom hos 374 patienter som registrerades genom systematisk sökning varierade inte nämnvärt mellan dem som jag diagnostiserade som cEDS respektive hEDS, och inte ens de 8 patienter som identifierades som möjliga vaskulära vEDS genom att de hade mutationer i kollagen typ III-genen var särskiljande (aneurysm som förekom hos en av de 8 patienterna hade samma låga frekvens i andra EDS-kategorier). Viktigast är begreppet EDS-spektrum, som enligt min mening bäst representeras av AAD.
Att erkänna symtomkomplexet AAD, som orsakas av EDS och besläktade bindvävsdysplasier, kommer att förhindra sorgliga patientresor från specialist till specialist, symtomdiagnoser som yrsel, svimning, fibromyalgi eller kronisk trötthet, och tragiskt lidande när många förebyggande strategier och behandlingar är tillgängliga. Dessutom förklarar erkännandet av dysautonomi den ångest som POTS ger upphov till och den depression som ofta åtföljer kronisk smärta eller aktivitetsbegränsning , beteendeskillnader som kan erkännas som konsekvenser av medicinsk snarare än psykiatrisk sjukdom.
Genetiska tester stöder konceptet articulo-autonomisk dysplasi
Med stöd av att de karaktäriseras som ärftliga sjukdomar i bindväv , visade sig de mer extrema formerna av EDS och besläktade sjukdomar uppvisa autosomal dominant nedärvning, vilket innebär att de drabbade individerna har en normal och en onormal form av genen (allelen). Dominant nedärvning är logiskt eftersom defekt protein som produceras av den onormala allelen, analogt med en skev tegelsten, skulle blandas med normala tegelstenar från den andra allelen och skapa en ojämn struktur. Att erkänna leden eller artikulationen som en struktur med flera komponenter, som begränsas av överliggande hud, stöds av inre ben och brosk och verkar genom inneboende nerver, muskler, senor, ledfodring och kärl, innebär i sin tur att någon av dessa delar kan påverkas och ge upphov till dysplasi, en vävnadsstörning som skiljer sig från de missbildningar som orsakas av andra genetiska störningar. Utvecklingen av DNA-kloning och sekvensering skulle bekräfta att autosomal dominant nedärvning fungerar och att gener som ändrar något element i ledstrukturen kan orsaka symtombilden för AAD.
Genetisk testning började med fokus på en ”kandidatgen” som valdes ut på grund av patientens symtom, vilket illustreras av fibrillin-1 gentestning för Marfans syndrom . Genom att sekvensera genens DNA AGCT-bokstäver, ”texten” av nukleotider, kunde man avslöja substitutioner eller ”skrivfel” som ändrade aminosyran i det kodade proteinet och dess avsedda funktion. Sedan kom den nya tekniken som kallas NextGen eller snabb parallell DNA-sekvensering, vilket kan liknas vid att läsa alla sidor i en bok på en gång i stället för från början till slut . Detta gjorde det möjligt att sekvensera gengrupper (paneler) som de som orsakar kardiomyopati eller bindvävsdysplasi eller till och med alla 23 000 gener i varje mänsklig cell (vårt genom) på 2-3 månader . Sekvensering av de proteinkodande eller exoniska genregionerna, kallad whole exome sequencing (WES), kan beställas från många företag och i min praktik har hälften av mina patienter fått den för mindre än 200-300 US-dollar (listpriset är 9000 US-dollar och det belopp som faktureras till försäkringen är cirka 20 000 US-dollar).
Och även om tolkningen av de i genomsnitt 12 000 DNA-varianter som beskrivs i WES förblir ett problem, som minskas av en växande epidemiologi av varianter i normal- och sjukdomspopulationer, är utbytet av genförändringar som bidrar till AAD-symtom bland 620 patienter som genomgår WES i min praktik över 60 %. Förutom över 80 mutationer i kollagengener, inklusive de i kollagen typ I som vanligtvis förknippas med osteogenesis imperfecta, kollagen typ V som ofta förknippas med klassisk EDS, kollagen typ VI, IX och XII som ursprungligen beskrevs i samband med myopatier och det fruktade kollagenet typ III som förknippas med vaskulär EDS, finns det återkommande mutationer i gener som påverkar hud-, nerv-, muskel-, fibrillär- och kärlkomponenter som stärker begreppet AAD, där en förändring av någon av ledkomponenterna ger upphov till det tecken- och symtomschema som beskrivs i tabell 1. Om EDS och relaterade sjukdomar som omfattas av AAD-konceptet nu erkänns kan det kliniska intrycket förstärkas med objektiva DNA-bevis som gör det möjligt att ge råd till patient och familj.
Diagnostik och behandling av hypermobilitetsstörningar/AAD
Att vara uppmärksam på huvudsymptomen ledsmärta/fragilitet och autonoma (tarm-, hjärt-, immunförsvarsstörningar) kan leda till snabbtest för hypermobilitet enligt figur 1. Tecknet tummen mot underarmen kan föregå hela Beighton-skalan och ge en poäng bilateralt för fingrar som pressas tillbaka bortom 90 grader på handen, röra tummen mot underarmen, hyperextension av armbågen med utsträckta armar, hyperextension av knäna för att bilda en bakåtriktad böjning av benet och 1 poäng för förmågan att röra handflatorna mot golvet . En kort undersökning av symtom som de som anges i tabell 1 kan bekräfta misstanken, eventuellt med hjälp av några av de bedömningar som anges i den nedre delen av tabell 1 som en del av de fysiska undersökningarna. Observera att alla AAD-patienter inte är långa eller smala och att en betydande del av dem kommer att vara överviktiga på grund av aktivitetsbegränsningar på grund av smärta och trötthet. Många kommer att ha elastisk hud, vilket undersöks genom att dra upp veck runt käken eller på underarmen, särskilt när dessa är tunna, ytliga veck av epidermis. Uppbyggnad av vit vävnad eller keloidbildning runt ärr är vanligt, och bedömning av Beighton eller andra hypermobilitetsmanövrer som anges i tabell 1 kan vara till hjälp. Man kan notera en nedsjunken hållning med framåtriktad nackkurva, skolios och lordos tillsammans med platta fötter eller att man går med tåna utåt (mer än inåt) när man går. Muskelstyrkan kommer att vara nedsatt hos vissa patienter tillsammans med dålig balans vid tandemgång, men en allmänläkare med begränsad tid behöver bara ta hand om ett fåtal av dessa fysiska tecken; resten kommer att fastställas på remiss.
Det viktigaste kommer att vara ortopeder som är bekanta med artros och medföljande förändringar i EDS som degenerativa ledförändringar, försämrat brosk, plicaband och områden med döda ben. En erfaren och konservativ ortopedist som inser de dåliga resultaten av operationer för att ”strama åt” eller ersätta leder är önskvärd. En kardiolog kan dokumentera POTS med hjälp av tilt-table-testet för ortostatiska förändringar i hjärtfrekvens och blodtryck och även utföra elektro- och ekokardiografiska undersökningar för att utesluta aortavidgning eller aneurysm. Neurologin kan bedöma vanlig migrän eller daglig huvudvärk, Chiari-deformationen som uppstår vid glidning och herniation av slapp hjärnvävnad, neuropati på grund av sammandragning i slappa leder och de muskelförändringar som anges i symtombeskrivningen fibromyalgi och som framhävs av frekventa myopatiska genförändringar, vilket ger upphov till en ny benämning, myopatiskt EDS . Allergologer som är bekanta med mastcellsaktivering och gastroenterologer som kan dokumentera låg tarmmotilitet är värdefulla tillägg till subspecialistteamet, och ingen är mer värdefull än primärläkaren när det gäller att hantera de många rekommendationer och mediciner som kan behövas. Även om det kan tyckas märkligt att komma från en genetiker tycker jag att de subspecialitetsutvärderingar som leder till terapi värderas högre än genetisk utvärdering och testning, men det senare kan vara till hjälp för att avdramatisera antaganden om psykiatrisk sjukdom .
Behandlingen börjar med ett träningsprogram som kan gynna både artrit på grund av ledhypermobilitet och dess reflexmässiga dysautonomi. Uppmärksamhet på ledvärk (växtvärk uppstår på natten) eller skada/skelettdeformitet kan instifta förebyggande åtgärder genom att göra patienten medveten om sitt tillstånd (notera i tabell 1 att många inte känner igen sin flexibilitet som onormal). Slitageskador kan förebyggas genom att man stödjer känsliga leder, använder midjeband för att stödja ryggen och väljer ledskyddande aktiviteter, t.ex. simning framför jogging. Patienterna bör förbli aktiva med måttlig tyngdlyftning och andra rimliga aktiviteter för att bygga upp musklerna runt lederna och förhindra cykler av inaktivitet som förvärrar kronisk trötthet eller fibromyalgi. Lämplig sjukgymnastik, smärtbehandling, träningsrådgivning och anekdotiska läkemedel som glukosamin/chondroitinpreparat har hjälpt många, men avslappningsmedel som tar bort muskelskyddet för lederna och steroidinjektioner som maskerar skador, främjar osteoporos/degeneration och endast ger tillfällig lindring bör undvikas.
Den viktigaste grunden för behandling av dysautonomi är hydrering (8-10 glas vätska per dag), salt (frånvarande labil hypertoni) och en proteinrik kost, som alla syftar till att öka blodets vätskevolym och förbättra den cerebrala cirkulationen. Ytterligare näringsbehandling kan omfatta C-vitamin (2 g per dag) D-vitamin (>2000 enheter per dag, mer än de 400-800 i multivitaminer), vitamin B12 (1-2,5 mg per dag). Låg tarmmotilitet eller gastropares hjälper man genom att undvika vätska före måltiderna, mindre och mer frekventa måltider, probiotika, enzympreparat och, enligt uppgift från många, mindre gluten och mejeriprodukter som är vanliga irriterande ämnen. Övningsprogram på ryggläge, biofeedback och många läkemedelsalternativ finns tillgängliga hos kardiologer för POTS (t.ex. betablockerare, Midodrine, Florinef), hos neurologer eller experter på smärtlindring (t.ex. Gabapentin, Lyrica) eller hos allergologer som kan tillhandahålla antihistaminprotokoll för att undertrycka mastcellerna (t.ex. Zyrtec, Zantac, Cromolyn, Singulair). Fördelarna med genomiska tester som WES som går utöver de gener som är kända för att orsaka bindvävsdysplasi är nya fynd, t.ex. de muskelrelaterade gener som nu är kända för att orsaka EDS, och dokumentation av tillfälliga eller sekundära fynd, t.ex. genmutationer för bröst- och äggstockscancer. Att hitta gener som är förknippade med allvarligare symtom gör det möjligt för patienterna att känna till riskerna för avkomman och att välja strategier för prenatal eller preimplantatorisk genetisk diagnostik som är förenliga med deras värderingar.
Fallbeskrivning
Eve kom till sin läkare när hon var 18 år gammal, yr, svimfärdig, orolig och magsjuk, så utmattad att hon drog sig tillbaka på fredagskvällar för att vila hela helgen. Hon hade varit frisk, aktiv inom gymnastik och cheerleading, en modig och smart tjej vars uthållighet trots skador fick sina tränare att applådera. Sedan kom influensan som hon aldrig återhämtade sig från, illamående så svårt att hon inte kunde äta, viktnedgång, svimning på lektionerna, uppvaknande på natten med ett bultande hjärta, uppvaknande på morgonen utan återhämtande sömn. Eve besökte många läkare, till och med akuten när hjärtklappning och panik tog andan ur henne. De sa att det inte var något fel, bara stress, och att allt var i hennes huvud. Hon lugnade sig men blev inte botad av piller, arbetet gick åt pipan på grund av trötthet och förvirring och hon undrade om hon höll på att bli galen. Hon kände inte igen sin mjuka och sköra hud, sina hårda och kraftiga menstruationer, sin dagliga huvudvärk och, mest slående av allt, sin hypermobilitet som hon och hennes familj trodde var normal: ”Ja, vi är böjda och har dubbla leder, är inte alla det?”. Eve skulle ha svarat om hon hade blivit tillfrågad.
Turligtvis sökte Eve en ny primärläkare som kunde dokumentera många av de tecken och symtom som anges i tabell I. Eve hade tidig kolik och reflux med svårigheter att amma, var mycket flexibel, kallades av sin mamma för den ”benlösa” bebisen, och när hon blev äldre satt hon på knäna med benen utsträckta bakom sig (W-positionen) och sa att det var bekvämare. Hon var till en början långsam att gå, ramlade ofta och verkade klumpig, men var snart aktiv och klättrade överallt, och fick sina föräldrar att rycka till när hon satte en fot runt hennes hals eller gjorde spagat med lätthet. Men när hon började gå i grundskolan började hon klaga på artralgi, benen gjorde ont på natten och knäna och fotlederna var besvärliga efter aktivitet. Hennes barnläkare försäkrade föräldrarna om att hon bara hade ”växtvärk”, men de blev mer oroliga när hon försökte spela fotboll och hela tiden vred om på fotlederna, en stukning krävde en känga i två månader efter att en stressfraktur konstaterats.
Eve var tvungen att sluta med fotbollen, men upptäckte att hon utmärkte sig i gymnastik när hon blev äldre, och valde golvarbeten på grund av dålig balans på bjälken och en tendens att hennes axel drogs ut på de parallella stängerna. Hon märkte att hennes leder hoppade i samband med rörelser och hoppade ofta på dem med flit eftersom det verkade lindra spänningar och smärta. Hon provade på cheerleading och lyckades bra med manövrer, men upptäckte att hon var för känslig för värme. De andra tjejerna kallade henne ”tomatansikte” eftersom hon blev överhettad under rutinerna, och hon blev lätt trött och behövde ofta en dags vila efter träningen. Hennes matsmältningsproblem återuppstod med frekventa förstoppningar och magsmärtor, hon kände sig ofta uppblåst och behövde laxermedel. I sjätte klass missade hon skolan på grund av sina tarmproblem och ledvärk och kände sig ofta trött och yr. Hon kände sig inte utvilad på morgonen och tog ofta tupplurar på helgerna, vilket var ett avsteg från hennes tidigare aktiva och entusiastiska livsstil. Hon kände sig ofta orolig, utan att kunna spåra det till någon specifik skol- eller familjestress, och började få huvudvärk på morgonen som hon trodde berodde på hennes dåliga sömn. Hennes föräldrar tog henne återigen till barnläkaren och fick återigen höra att det inte var något fel utan vanlig skolstress.
När hon var 14 år hade Eve en svår influensaliknande sjukdom och missade skolan i en månad med svår trötthet, huvudvärk, illamående, kräkningar och viktnedgång. Hon hade fruktansvärd yrsel och viss bröstsmärta och kände att hennes hjärta bultade ibland (hade hjärtklappning). Hennes föräldrar sökte råd hos en annan barnläkare som skickade Eve till en kardiolog som trodde att hon hade takykardi på grund av stress och ångest i samband med hennes sjukdom. Han gav Eve propranolol, en betablockerare, för att sänka hennes hjärtfrekvens, men hon blev ännu mer trött och var nu sängliggande en stor del av dagen. Hennes ledvärk ökade efter sjukdomen och hon stannade kvar i sängen större delen av dagen, kände sig ledsen och blev orolig när hennes föräldrar försökte ta med henne ut eller ta med henne på en biltur. De tog henne tillbaka till den andra barnläkaren som hänvisade henne till psykiatrin, och Zoloft påbörjades för att hjälpa mot ångest och depression. Efter några dagar fick Eve utslag med nässelutslag och var tvungen att sluta med medicinen. Hon fortsatte att ha yrsel och träffades av ytterligare två specialister, en gastroenterolog som trodde att hon kunde ha celiaki och en öron-näsa-halsspecialist som diagnostiserade yrsel och skickade henne till neurologin. Hon berättade för den läkaren om huvudvärk som utgick från bakhuvudet, och en MRI-studie av huvudet i liggande ställning gjordes som var normal.
Med tanke på den troliga AAD-diagnosen från hennes primärläkare inleddes en annan omgång remisser till subspecialiteter, inklusive neurologi där en MRI-studie av huvudet i liggande ställning visade att det fanns en Chiari-deformation i form av herniation. En kardiolog dokumenterade ett onormalt tilt-table-test tillsammans med låg tarmmotilitet och bevisade att hennes ångest och stress-trötthet berodde på dysautonomi. Strategier för näringshydrering, salt och proteinrika produkter samt behandling med betablockerare och antihistaminer förbättrade hennes trötthet och tarmsymtom och minskade hennes huvudvärk. Det beslutades att Chiari-herniationen på 4-5 mm inte var tillräcklig för att kräva operation, och hennes psykiatriska behandling erkändes som behandling av sekundära symtom på en medicinsk störning. Några av hennes subspecialister, som var skeptiska till hennes kliniska diagnos, övertygades av genetiska tester som dokumenterade en förändring i en gen för alfa-1-kedjan av typ V-kollagen, förkortat COL5A1, ett vanligt fynd hos patienter med EDS. Eve hade ärvt mutationen från sin mor, som hade haft mildare AAD-symtom och nu kunde hantera dem bättre.
Under flera månader återfanns Eves hälsa och vitalitet och hon ”fick sitt liv tillbaka”, som hon och hennes föräldrar skulle säga. Hon hade fortfarande ledvärk, ångest och trötthet ibland, men ”kloka övningar” för att bygga upp muskelskydd runt lederna, näringsmetoder för dysautonomi och biofeedbackmetoder för att förebygga ångest förändrade hennes liv. Alla dessa behandlingar berodde på att hennes husläkare erkände dem, som nu kunde samordna vården för Eve och hennes mamma och tillhandahålla resurser för att förstå både EDS och de underliggande genförändringarna. Övergång till nästa generation var möjligheten att informera Eve om hennes 50-procentiga risk att föra sin COL5A-mutation vidare och om vissa risker för tidig förlossning och blödning efter födseln vid framtida graviditeter .
- Remvig L, Jensen DV, Ward RC (2007) Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: review of the literature. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) Ehlers-Danlos syndrom. I: Ehlers-Danhlers syndrom (Ehlers-Danhlers syndrom): Connective Tissue and its Heritable Disorders. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomia in the joint hypermobility syndrome. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lärdomar om smärtlindring – en personlig erfarenhet efter examen. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) En liten molekyl för en stor sjukdom. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Kapitel 11: Gen- och genomsekvensering: Tolkning av genetisk variation på nukleotidnivå. In: Human Chromosome Variation: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing–are we there yet? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Benign joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Muskuloskeletala besvär i samband med generaliserad ledhypermobilitet. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Presymptomatisk och preimplantatorisk genetisk diagnostik: Neurologi, NextGenetics och nästa generation. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Behandling av smärta och trötthet vid hypermobilitetssyndrom i lederna (även kallat Ehlers-Danlos syndrom, hypermobilitetstyp): principer och förslag till en multidisciplinär strategi. Am J Med Genet A 158:
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Multidisciplinär behandling av funktionsnedsättning vid Ehlers-Danlos syndrom hypermobilitetstyp/ hypermobilitetssyndrom: en pilotstudie med en kombination av fysisk och kognitiv beteendeterapi på 12 kvinnor. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exome analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am J Med Genet A 164A: 1209-1212.