Schlüsselwörter
Hypermobilität, Bindegewebsdysplasie, Ehlers-Danlos-Syndrom, Beighton-Skala, Dysautonomie, posturales orthostatisches Tachykardiesyndrom, Reizdarmsyndrom, Mastzellaktivierungsstörung, artikulo-autonome Dysplasie
Einführung
Völlig unterschätzt und tragischerweise falsch behandelt werden die 20 % der Frauen und 10 % der Männer, deren Gewebe überdurchschnittlich flexibel und zerbrechlich ist. Es gibt nur wenige Möglichkeiten der Vorbeugung und Behandlung, die über die Erkennung der Hypermobilität hinausgehen. Diese lässt sich leicht feststellen, indem man den Patienten bittet, den Daumen gegen den Unterarm zu drücken (Abbildung 1). Dieses verräterische Zeichen kann den Weg zu einer detaillierteren Untersuchung weisen, die gut in den Bereich des Allgemeinmediziners fällt, dessen breites klinisches Wissen gut zu den Multisystem-Problemen passt, die durch laxes Bindegewebe entstehen können. Hier gebe ich einen Überblick über das Wesen von Hypermobilitätsstörungen, zeige auf, dass allgemeines statt fachfremdes Wissen zu lebensverändernden Therapien für diese Patienten führen kann, und zeige, wie Gentests symptomatische Beschreibungen wie Fibromyalgie oder chronisches Müdigkeitssyndrom in das erkennende Licht der Präzisionsmedizin rücken.
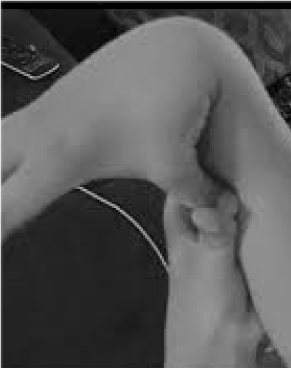
Abbildung 1. Hypermobilität.
Es müssen mehrere Hindernisse überwunden werden, bevor Patienten mit Hypermobilität in den Genuss der Vorteile der Allgemeinmedizin kommen können. Die erste betrifft den bahnbrechenden, aber kurzsichtigen Fokus der Dermatologen Ehlers und Danlos, die unglaubliche und exotische Hautdehnungen als irreführendes Symbol für Bindegewebslaxitätsstörungen prägten. Ein zweites Hindernis ist die zunehmende Kluft zwischen praktischer und genetischer Medizin, wobei sich erstere auf häufige Krankheiten konzentriert, während letztere bis vor kurzem spezialisierte Technologien zur Beschreibung seltener Krankheiten verwendete. Die obskuren Eponyme mit ihren entmutigenden Subtypen und Fachbegriffen sind wie kleine Provinzen eines wenig einladenden fremden Landes, für das sich kein Weltreisender die Zeit nimmt, eine Karte zu erstellen. Um diesen Hindernissen zu begegnen, ist ein praktischer Ansatz für Hypermobilitätsstörungen erforderlich, der sich auf die Erkennung durch Anamnese und körperliche Untersuchung konzentriert und die technischen Einzelheiten der Eponyme, Subtypen und genetischen Tests den Spezialisten überlässt, die nach dem wichtigen ersten Schritt der Verdachtsdiagnose kontaktiert werden. Nach der Erkennung und Überweisung bleibt der flexible Patient von seinem Hausarzt abhängig, denn nur er kann Empfehlungen und Testergebnisse in die allgemeine medizinische Versorgung einbeziehen.
Terminologie
Eine eingeschränkte Beweglichkeit an mehreren Gelenkstellen tritt bei einer Vielzahl von Krankheiten auf, die von Chromosomenstörungen wie dem Down-Syndrom bis hin zu Erkrankungen wie dem Ehlers-Danlos-Syndrom (EDS) reichen, das für seine Hypermobilität bekannt ist. McKusick machte diese Kategorie von Erkrankungen, die für ihre Gewebelaxität bekannt sind, populär, indem er sie als vererbbare Bindegewebsstörungen zusammenfasste, darunter das Marfan-Syndrom, auf das er sich als Kardiologe konzentrierte. 1977 organisierte Dr. Peter Beighton, der für seine Skala zur Messung der Flexibilität bekannt ist, die dazwischen liegende Literatur, indem er 6-7 EDS-Typen postulierte, die nun in den jüngsten Leitlinien vereinfacht als klassisches, vaskuläres und hypermobiles EDS bezeichnet werden. Inzwischen ist klar, dass sich die Symptome dieser Typen untereinander und mit verwandten Erkrankungen wie Osteogenesis imperfect (mit zahlreichen Frakturen), Marfan-Syndrom (große Gefäßdilatation/-dissektion), Stickler-Syndrom (Augen- und Arthritisveränderungen) und vielen anderen erheblich überschneiden.
Anstatt sich mit einzelnen Krankheiten zu befassen, halte ich es für sinnvoller, sich auf zwei große Krankheitsprozesse von Bindegewebsdysplasien zu konzentrieren, die durch die vielen in Tabelle 1 aufgeführten Symptome veranschaulicht werden. Der erste ist der abnutzungsbedingte Gelenkschmerz (Osteoarthritis), der sich von den geschwollenen, geröteten Gelenken der entzündlichen Arthritis (Autoimmun- oder entzündliche Arthritis) unterscheidet, die bei rheumatoider Arthritis, Lupus, Sjögren-Syndrom oder Morbus Bechterew auftritt. Die zweite Ursache ist das Ungleichgewicht des autonomen Nervensystems (Dysautonomie), das durch elastische Blutgefäße bei Hypermobilitätsstörungen verursacht wird. Die dehnbaren Gefäße führen zu Blutansammlungen im Becken und in den Beinen, was einen niedrigeren Blutdruck und Schwindel/Schwäche beim Stehen zur Folge hat (orthostatische Hypotension). Mit der Zeit aktiviert der verminderte Blutfluss zum Kopf den sympathischen Arm des autonomen Nervensystems (den Adrenalinarm „Flucht oder Kampf“), was zu mehreren Problemen führt: 1) posturales orthostatisches Tachykardiesyndrom (POTS) mit Tachykardie, Angstzuständen, chronischer Müdigkeit und „Brain Fog“ (Intervalle mit verminderter Konzentration und Gedächtnisleistung), 2) Reizdarmsyndrom aufgrund der Hemmung der Darmmotilität (parasympathische Suppression) mit Verstopfung im Wechsel mit Durchfall, Blähungen, Reflux, Magenschmerzen, Dysphagie, Übelkeit und 3) Mastzellaktivierungsstörung mit vorübergehenden Hautausschlägen, Nesselsucht/reaktiver Haut, Asthma/Atemnot und Nahrungsmittel-/Medikamentenunverträglichkeiten. Ich habe einen umfassenderen Begriff entwickelt, der die Erkennung jeder Hypermobilitätsstörung erleichtert und die beiden Hauptfolgen der Gelenk-/Gewebelaxität anerkennt: artikulo-autonome Dysplasie oder Gelenk-Ermüdungskomplex.
Tabelle 1. Typische Befunde und ihre Häufigkeit bei Hypermobilitätssyndromen. *Aus 320 weiblichen und 54 männlichen Patienten über 12 Jahren, die 2017 mittels detaillierter Fragebögen erfasst wurden; TMJ, Temporomandibulargelenk; POTS, posturales orthostatisches Tachykardiesyndrom. Symptomliste teilweise angepasst aus Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
GESCHICHTE (Prozentangaben)*
Säuglingsalter: Koliken (28), Schwierigkeiten beim Füttern (19,4); ungeschickt, viele Stürze (29,1)
Kind/Jugendliche: Langsame Gewichtszunahme (12); auf Beweglichkeit achten (55,6); Gelenktricks vorführen (53,6)
frühe Gelenkschmerzen (42,1); Teenager-Aktivitäten durch Schmerz-Verletzung eingeschränkt (55,4); frühe Brille (46,8)
Kieferorthopädie (61,4%) Zahnfleischerkrankungen (11,3); schlechter Zahnschmelz/viel Karies (31,6)
Gelenkprobleme: Knacken (81,6); Subluxationen (62); Kiefergelenkknacken, Einklemmen, Schmerzen (51,2)
Schmerzen in mindestens 2 Gelenken (80,3); Gelenkoperationen (33); viele Verstauchungen (26); Frakturen >2 (31,5)
Bandscheibenvorfall (29,3)
Skelettprobleme: Skoliose (31); Pectus (2,2); Gang – einwärts/auswärts gehen (9,3); Plattfüße (15,5)
Haut: Leichte Blutergüsse (61); dehnbare Haut (15); samtig (18); ungewöhnliche Narben (36,3)
Striemen vor der Schwangerschaft (73); langsame Heilung (32,4)
Genitourinär (Frauen): Menorrhagie (61,2); Endometriose (11,3); Eierstockzysten (55)
Polyzystisches Ovarialsyndrom (8,5); Blasenprobleme (46,1); Leisten-/Femoralhernie (11,5)
Neuromuskulär: Migräne (51,7); tägliche Kopfschmerzen (62,3); benötigte Kopfschmerzmedikamente (45)
Chiari-Deformation (10,6); neurochirurgische Eingriffe (2,9); Taubheitsgefühle/Kribbeln (48,7); Gleichgewichtsstörungen (48,6)
neuropathische einschießende Schmerzen (37,3); Muskelschmerzen/Krämpfe (51,7); Muskelschwäche (32,4)
GI/Darm: Verstopfung-Durchfall (67,1); Blähungen-Reflux-Magenschmerzen (57,3)
Gallenblasenprobleme (12,9); Schluckbeschwerden (23); häufige Übelkeit (44,9)
Herz: Mitralklappenprolaps (8,7); Aneurysma (0,9); Herzrhythmusstörungen außer Tachykardie (6,8)
Punkte: Schwindel beim Stehen (69,8); Synkope (34,5); chronische Müdigkeit (81,8); Schlafstörungen (43,5)
Gehirnnebel (zeitweise Gedächtnis- oder Konzentrationsschwäche-70,8); Hitze-/Kälteempfindlichkeit (77,5); abnormales Schwitzen (25); Tachykardie (72,8); Angst-Panikattacken (61,3); Salzsucht (51,2)
Immunerkrankungen/Mastzellen: Vorübergehende Hautausschläge (33,4 %); Nesselsucht/reaktive Haut (45,9 %); Asthma/Atemnot (39,3); Nahrungsmittel-/Medikamentenunverträglichkeiten (61,5 %)
Labor/Bildgebung: Niedrige Knochendichte (2,1); niedriges Vitamin D (28,7); niedriges Vitamin B12 (2,7); Hypothyreose (11,5); niedriges Ferritin (1,7); niedriges Eisen (6,7); erhöhte antinukleäre Antikörper (7.1)
Durchschnittliche Anzahl der Befunde von 80: Frauen >12 (35,4 Befunde) Männer >12 (21,5 Befunde)
Physikalisch (Prozent)
Körperbau: Hochgewachsen >90. Perzentil (55,2); schwerer BMI >28 (21,3); dünner BMI <19 (14,2)
Marfanoider Habitus (44); Arachnodaktylie (46,4); Walker-Murdoch Zeichen vorhanden (40,2)
Gesicht: Lang (25.7); enges Untergesicht (5.4); blau-graue Sklera (1.6); hoher Gaumen (53.5)
Haut: Weich (81,4); durchscheinend (23,8); elastisch-liftig >1 Zoll Falte um den Kiefer auf der Mitte des Unterarms (51,3)
Falten bestehen aus der äußeren (epidermalen) Schicht (35.4); ungewöhnliche Narben (42,1%)
Beighton-Score: 0-3 Punkte (8,4); 4-6 Punkte (32,6); 7-8 Punkte (38,8); 9 von 9 Punkten (20,2)
Weitere Manöver: Hände über den Schultern hinter dem Rücken verschränken (68)
Gebetszeichen nach oben hinter dem Rücken (71,3); Hand um den Rücken führen, um den Nabel zu berühren (19,5)
Skelettale: Nackenkyphose (41,2); Skoliose (22,3); Lordose (32,8); Plattfüße (39,6)
Gang – einwärts oder auswärts gehen (28,3)
Neurologisch: Muskelschwäche (11,2); verminderte Muskelmasse (8,8)
Gleichgewichtsstörungen beim Tandemgang (33,4)
Durchschnittliche Anzahl Befunde von 40: Frauen >12 (17,9 Befunde) Männer >12 (15.7 Befunde)
Artikulo-Autonomische Dysplasie (AAD)
Ein Begriff wie „artikulo-autonomische Dysplasie“ oder AAD kann den Ansatz für Bindegewebslaxität/Hypermobilitätsstörungen vereinheitlichen und drei Problemen entgegenwirken, die ihre Anerkennung erschweren: 1) Viele Ärzte und Genetiker betrachten Bindegewebsdysplasien als seltene und extreme Krankheiten, obwohl sie in Wirklichkeit etwa 1 % der Bevölkerung betreffen. 2) Die EDS-Diagnoserichtlinien vereinfachen zwar die wichtigsten EDS-Typen wie klassisches, vaskuläres und hypermobiles EDS, verwirren aber immer noch, indem sie sich auf viele kleinere Typen beziehen, die ein sich überschneidendes Symptomspektrum bilden, und 3) die fast obligate Dysautonomie, die EDS und verwandte Störungen begleitet (Tabelle 1), wird als diagnostisches Kriterium weggelassen, was der Beschreibung eines Elefanten ohne Erwähnung seines Rüssels entspricht. AAD bezeichnet die unvermeidliche physiologische Verbindung zwischen lockerem Gewebe und Dysautonomie in der gleichen Weise, wie die metabolischen oder hepatorenalen Syndrome beschreiben, wie Fettleibigkeit zu Lebererkrankungen/Diabetes oder Leberversagen zu Nierenerkrankungen führt.
Wie die meisten medizinischen Muster oder Syndrome, einschließlich EDS, ist AAD ein Symptomkomplex mit vielen zugrunde liegenden Ursachen. „Articulo“ bezieht sich auf das Gelenk, wobei anerkannt wird, dass die Erkrankung jeder Gelenkkomponente – Haut, Nerven, Muskeln, Knochen, Bindegewebe, Blutgefäße – ein ähnliches Muster der Laxheit des Gelenkgewebes mit autonomem Ungleichgewicht verursacht. Es kommt zu Hautbrüchigkeit, verminderter Knochendichte mit Anfälligkeit für Frakturen, Muskelschwäche mit Schmerzen (Myalgie), Neuropathie mit Taubheitsgefühlen/Kribbeln, Karpaltunnel oder regionalen Schmerzsyndromen, autonomen Veränderungen einschließlich Tachykardie, Angstzuständen, nicht erholsamem Schlaf, Darmimmotilität, Nesselsucht/Ausschlag, Nahrungsmittel- und Medikamentenunverträglichkeiten, die alle in unterschiedlichem Ausmaß auch innerhalb bestimmter „Typen“ von EDS auftreten. Ich habe keine großen Unterschiede zwischen Patienten gefunden, die zu bestimmten EDS-Typen passen, wie z. B. häufigere Hautveränderungen bei Typ I klassischem cEDS und größere Gelenkflexibilität bei Typ III hypermobilem hEDS. Die Häufigkeit der Symptome bei 374 Patienten, die durch systematische Abfragen erfasst wurden, unterschied sich nicht signifikant zwischen denjenigen, die ich als cEDS und hEDS diagnostizierte, und selbst die 8 Patienten, die aufgrund von Mutationen des Kollagen-Typ-III-Gens als mögliches vaskuläres vEDS identifiziert wurden, unterschieden sich nicht voneinander (Aneurysmen, die bei einem der 8 Patienten auftraten, waren in anderen EDS-Kategorien ebenso selten). Am wichtigsten ist das Konzept eines EDS-Spektrums, das meiner Meinung nach am besten als AAD dargestellt wird.
Die Anerkennung des AAD-Symptomenkomplexes, der durch EDS und verwandte Bindegewebsdysplasien verursacht wird, wird traurige Patientenreisen von Spezialist zu Spezialist, symptomatische Diagnosen wie Schwindel, Ohnmacht, Fibromyalgie oder chronische Müdigkeit und tragisches Leiden verhindern, obwohl viele Präventionsstrategien und Behandlungen verfügbar sind. Darüber hinaus erklärt die Anerkennung der Dysautonomie die Angst vor POTS und die Depression, die häufig mit chronischen Schmerzen oder Aktivitätseinschränkungen einhergeht, Verhaltensunterschiede, die als Folgen einer medizinischen und nicht einer psychiatrischen Erkrankung erkannt werden können.
Genetische Tests unterstützen das Konzept der artikulo-autonomen Dysplasie
Durch ihre Charakterisierung als vererbbare Bindegewebsstörungen wurde festgestellt, dass die extremeren Formen von EDS und verwandten Störungen einen autosomal-dominanten Erbgang aufweisen, was bedeutet, dass die betroffenen Personen eine normale und eine abnorme Form des Gens (Allel) haben. Die dominante Vererbung ist sinnvoll, weil sich das vom abnormen Allel produzierte defekte Protein, ähnlich wie ein verzogener Ziegelstein, mit den normalen Ziegelsteinen des anderen Allels vermischen und eine wackelige Struktur bilden würde. Die Anerkennung des Gelenks oder der Artikulation als eine Mehrkomponentenstruktur, die durch die darüber liegende Haut eingeengt wird, von den inneren Knochen und Knorpeln gestützt wird und durch intrinsische Nerven, Muskeln, Sehnen, Gelenkauskleidung und Gefäße wirkt, bedeutet wiederum, dass jedes dieser Elemente beeinflusst werden kann, um eine Dysplasie zu erzeugen, eine Gewebestörung, die sich von den durch andere genetische Störungen verursachten Fehlbildungen unterscheidet. Die Entwicklung des DNA-Klonens und der Sequenzierung würde die Funktionsweise des autosomal-dominanten Erbgangs und die Fähigkeit von Genen, die irgendein Element der Gelenkstruktur verändern, das Symptommuster der AAD zu verursachen, bestätigen.
Genetische Tests begannen mit der Konzentration auf ein „Kandidaten“-Gen, das aufgrund von Patientensymptomen ausgewählt wurde, wie der Fibrillin-1-Gentest für das Marfan-Syndrom zeigt. Die Sequenzierung der DNA des Gens, der AGCT-Buchstaben, des „Textes“ der Nukleotide, konnte Substitutionen oder „Tippfehler“ aufdecken, die die Aminosäure des kodierten Proteins und seine beabsichtigte Funktion veränderten. Dann kam die neue Technologie namens NextGen oder schnelle parallele DNA-Sequenzierung, die dem Lesen aller Seiten eines Buches auf einmal und nicht von Ende zu Ende entspricht. Dies ermöglichte die Sequenzierung von Gengruppen (Panels) wie denjenigen, die Kardiomyopathie oder Bindegewebsdysplasie verursachen, oder sogar aller 23 000 Gene in jeder menschlichen Zelle (unserem Genom) in 2-3 Monaten. Die Sequenzierung der proteincodierenden oder exonischen Genregionen, die so genannte Ganz-Exom-Sequenzierung (WES), kann bei vielen Unternehmen bestellt werden und kostete in meiner Praxis bei der Hälfte meiner Patienten weniger als 200-300 Dollar (der Listenpreis liegt bei 9000 Dollar und der Betrag, der der Versicherung in Rechnung gestellt wird, beträgt etwa 20.000 Dollar).
Obwohl die Interpretation der durchschnittlich 12.000 DNA-Varianten, die durch WES detailliert erfasst werden, ein Problem bleibt, das durch eine wachsende Epidemiologie von Varianten in normalen und kranken Populationen verringert wird, liegt die Ausbeute an Genveränderungen, die zu AAD-Symptomen beitragen, bei 620 Patienten, die sich in meiner Praxis einer WES unterziehen, bei über 60 %. Zusätzlich zu den mehr als 80 Mutationen in Kollagengenen, einschließlich derjenigen in Kollagen Typ I, das üblicherweise mit Osteogenesis imperfecta assoziiert wird, Kollagen Typ V, das häufig mit klassischem EDS assoziiert wird, Kollagen Typ VI, IX und XII, die ursprünglich mit Myopathien beschrieben wurden, und das gefürchtete Kollagen Typ III, das mit vaskulärem EDS assoziiert wird, gibt es wiederkehrende Mutationen in Genen, die Haut-, Nerven-, Muskel-, fibrilläre und Gefäßkomponenten betreffen, die das Konzept der AAD verstärken, bei dem die Veränderung einer beliebigen Gelenkkomponente das in Tabelle 1 skizzierte Zeichen- und Symptommuster erzeugt. Die Erkennung von EDS und verwandten Störungen, die unter das AAD-Konzept fallen, kann nun den klinischen Eindruck durch objektive DNA-Beweise untermauern, die eine Beratung des Patienten und der Familie ermöglichen.
Diagnose und Therapie von Hypermobilitätsstörungen/AAD
Die Aufmerksamkeit für die Hauptsymptome Gelenkschmerzen/Fragilität und autonome Störungen (Darm, Herz, Immunsystem) kann zu Schnelltests für Hypermobilität führen, wie in Abbildung 1 dargestellt. Das Daumen-Unterarm-Zeichen kann der gesamten Beighton-Skala vorangestellt werden, wobei ein Punkt beidseitig vergeben wird, wenn die Finger in einem Winkel von mehr als 90 Grad an der Hand nach hinten gedrückt werden, wenn die Daumen die Unterarme berühren, wenn der Ellenbogen bei gestreckten Armen überstreckt wird, wenn die Knie überstreckt werden, um eine nach hinten gerichtete Beinbeuge zu bilden, und ein Punkt für die Fähigkeit, die Handflächen auf dem Boden zu berühren. Eine kurze Befragung zu Symptomen wie in Tabelle 1 kann den Verdacht bestätigen, eventuell unterstützt durch die Einbeziehung einiger der im unteren Teil von Tabelle 1 aufgeführten Bewertungen in die körperliche Untersuchung. Es ist zu beachten, dass nicht alle AAD-Patienten groß oder dünn sind und dass ein erheblicher Teil aufgrund von Aktivitätseinschränkungen durch Schmerzen und Müdigkeit übergewichtig ist. Viele haben eine elastische Haut, die durch Hochziehen der Falten um den Kiefer oder am Unterarm untersucht werden kann, insbesondere wenn es sich um die dünnen, oberflächlichen Falten der Epidermis handelt. Eine Ansammlung von weißem Gewebe oder Keloidbildung um Narben herum ist häufig, und die Bewertung von Beighton oder anderen in Tabelle 1 aufgeführten Hypermobilitätsmanövern kann hilfreich sein. Eine zusammengesackte Haltung mit nach vorne gekrümmtem Nacken, Skoliose und Lordose kann ebenso festgestellt werden wie Plattfüße oder das Absetzen der Zehen (mehr als das Eintreten) beim Gehen. Bei einigen Patienten ist die Muskelkraft vermindert und das Gleichgewicht beim Tandem-Gehen gestört, aber der Allgemeinmediziner, der nur wenig Zeit hat, braucht nur auf einige dieser körperlichen Anzeichen zu achten; der Rest wird durch Überweisung festgestellt.
Am wichtigsten sind Orthopäden, die mit der Arthrose und den begleitenden Veränderungen bei EDS vertraut sind, wie z. B. degenerative Gelenkveränderungen, sich verschlechternder Knorpel, Plicabänder und Bereiche mit totem Knochen. Ein erfahrener und konservativer Orthopäde, der die schlechten Ergebnisse von Operationen zur „Straffung“ oder zum Ersatz von Gelenken kennt, ist wünschenswert. Ein Kardiologe kann POTS mit Hilfe des Kipptisch-Tests für orthostatische Veränderungen der Herzfrequenz und des Blutdrucks dokumentieren und auch elektro- und echokardiographische Untersuchungen durchführen, um eine Aortenerweiterung oder ein Aneurysma auszuschließen. Die Neurologie kann häufige Migräne oder tägliche Kopfschmerzen, die Chiari-Deformation, die mit dem Verrutschen und der Herniation von schlaffem Hirngewebe einhergeht, die Neuropathie aufgrund von Einengungen in schlaffen Gelenken und die Muskelveränderungen, die durch die Symptombeschreibung Fibromyalgie bezeichnet werden und durch häufige myopathische Genveränderungen hervorgehoben werden, die den neuen Begriff des myopathischen EDS fördern, beurteilen. Allergologen, die sich mit der Aktivierung von Mastzellen auskennen, und Gastroenterologen, die eine niedrige Darmmotilität dokumentieren können, sind wertvolle Ergänzungen für das Team der Subspezialisten, und niemand ist wertvoller als der Hausarzt, wenn es darum geht, die vielen Empfehlungen und Medikamente zu verwalten, die möglicherweise erforderlich sind. Auch wenn es seltsam erscheinen mag, wenn es von einem Genetiker kommt, denke ich, dass die Beurteilungen der Subspezialitäten, die zu einer Therapie führen, höher zu bewerten sind als genetische Beurteilungen und Tests, aber letztere können hilfreich sein, um Annahmen über psychiatrische Erkrankungen zu entkräften.
Die Behandlung beginnt mit einem Übungsprogramm, das sowohl die Arthritis aufgrund der Gelenkhypermobilität als auch die reflexive Dysautonomie begünstigen kann. Die Aufmerksamkeit für Gelenkschmerzen (Wachstumsschmerzen treten nachts auf) oder Verletzungen/Skelettdeformationen kann eine Prävention einleiten, indem der Patient auf seinen Zustand aufmerksam gemacht wird (in Tabelle 1 ist zu beachten, dass viele ihre Flexibilität nicht als anormal erkennen). Abnutzungsschäden können verhindert werden, indem anfällige Gelenke gestützt werden, Hüftgurte zur Unterstützung des Rückens angelegt werden und gelenkschonende Aktivitäten wie Schwimmen dem Joggen vorgezogen werden. Die Patienten sollten mit moderatem Gewichtheben und anderen sinnvollen Aktivitäten aktiv bleiben, um die Muskeln um die Gelenke herum aufzubauen und so Zyklen der Inaktivität zu vermeiden, die chronische Müdigkeit oder Fibromyalgie verschlimmern. Angemessene Physiotherapie, Schmerzmanagement, Übungsanleitungen und anekdotische Medikamente wie Glucosamin/Chondroitin-Präparate haben vielen geholfen, aber Entspannungsmittel, die den Muskelschutz der Gelenke aufheben, und Steroidinjektionen, die Verletzungen maskieren, Osteoporose/Degeneration fördern und nur vorübergehend Linderung verschaffen, sollten vermieden werden.
Die Hauptpfeiler der Dysautonomie-Therapie sind Flüssigkeitszufuhr (8-10 Gläser Flüssigkeit pro Tag), Salz (bei labilem Bluthochdruck) und eine eiweißreiche Ernährung, die alle darauf abzielen, das Flüssigkeitsvolumen im Blut zu erhöhen und die zerebrale Durchblutung zu verbessern. Eine zusätzliche Ernährungstherapie kann Vitamin C (2 g pro Tag), Vitamin D (>2000 Einheiten pro Tag, mehr als die 400-800 in Multivitaminen) und Vitamin B12 (1-2,5 mg pro Tag) umfassen. Eine niedrige Darmmotilität oder Gastroparese wird durch den Verzicht auf Flüssigkeiten vor den Mahlzeiten, kleinere und häufigere Mahlzeiten, Probiotika, Enzympräparate und, wie viele berichten, durch den Verzicht auf Gluten und Milchprodukte, die häufige Reizstoffe sind, unterstützt. Übungsprogramme in Rückenlage, Biofeedback und viele medikamentöse Optionen sind bei Kardiologen für POTS (z. B. Betablocker, Midodrine, Florinef), bei Neurologen oder Schmerzspezialisten (z. B. Gabapentin, Lyrica) oder bei Allergologen erhältlich, die Antihistamin-Protokolle zur Unterdrückung von Mastzellen (z. B. Zyrtec, Zantac, Cromolyn, Singulair) anbieten können. Die Vorteile von Genomtests wie WES, die über die Gene hinausgehen, von denen bekannt ist, dass sie Bindegewebsdysplasie verursachen, sind neue Erkenntnisse wie die muskelbezogenen Gene, von denen jetzt bekannt ist, dass sie EDS verursachen, und die Dokumentation zufälliger oder sekundärer Befunde wie die Genmutationen bei Brust- und Eierstockkrebs. Die Entdeckung von Genen, die mit schwereren Symptomen in Verbindung stehen, ermöglicht es den Patienten, die Risiken für ihre Nachkommen zu kennen und Strategien der pränatalen oder Präimplantationsdiagnostik zu wählen, die mit ihren Werten vereinbar sind.
Falldarstellung
Eve kam im Alter von 18 Jahren zu ihrem Arzt, schwindlig, schwach, ängstlich und so erschöpft, dass sie sich freitagabends zurückzog, um das ganze Wochenende zu ruhen. Sie war gesund, aktiv in Gymnastik und Cheerleading, ein tapferes und aufgewecktes Mädchen, dessen Ausdauer trotz Verletzungen den Beifall ihrer Trainer fand. Dann kam eine Grippe, von der sie sich nie wieder erholte, eine so starke Übelkeit, dass sie nichts essen konnte, Gewichtsverlust, Ohnmachtsanfälle im Unterricht, nächtliches Aufwachen mit Herzklopfen, morgendliches Aufwachen ohne erholsamen Schlaf. Eve war bei vielen Ärzten, sogar in der Notaufnahme, als Herzklopfen und Panik ihr den Atem raubten. Sie sagten, es sei alles nur Stress, alles in ihrem Kopf. Die Tabletten beruhigten sie, aber heilten sie nicht, die Arbeit scheiterte an Müdigkeit und Verwirrung, und sie fragte sich, ob sie verrückt geworden war. Unerkannt blieben ihre weiche und zerbrechliche Haut, ihre harte und starke Periode, ihre täglichen Kopfschmerzen und vor allem ihre Hypermobilität, von der sie und ihre Familie annahmen, sie sei normal: „Ja, wir sind biegsam und doppelgelenkig, nicht wahr?“ Eve hätte geantwortet, wenn man sie gefragt hätte.
Glücklicherweise suchte Eve einen neuen Hausarzt auf, der viele der in Tabelle I aufgeführten Anzeichen und Symptome dokumentieren konnte. Eve hatte früh Koliken und Reflux mit Schwierigkeiten beim Stillen, war sehr biegsam, wurde von ihrer Mutter als „knochenloses“ Baby bezeichnet und saß, als sie älter war, auf den Knien, wobei sie die Beine hinter sich ausstreckte (die „W“-Position), weil sie meinte, das sei bequemer. Anfangs konnte sie nur langsam laufen, fiel häufig hin und wirkte unbeholfen, aber bald war sie aktiv und kletterte überall hin und ließ ihre Eltern zusammenzucken, wenn sie ihr einen Fuß um den Hals legte oder mit Leichtigkeit einen Spagat machte. Doch als sie in die Grundschule kam, begann sie über Gelenkschmerzen zu klagen. Ihre Beine taten ihr nachts weh, und ihre Knie und Knöchel machten ihr nach jeder Aktivität zu schaffen. Ihr Kinderarzt beruhigte ihre Eltern, dass es sich nur um „Wachstumsschmerzen“ handelte, aber sie machten sich mehr Sorgen, als sie versuchte, Fußball zu spielen und sich dabei immer wieder die Knöchel verdrehte; eine Verstauchung erforderte einen Stiefel für zwei Monate, nachdem eine Stressfraktur festgestellt worden war.
Eve musste mit dem Fußballspielen aufhören, aber sie fand heraus, dass sie sich beim Turnen auszeichnete, als sie älter wurde, und entschied sich für Bodenübungen, weil sie auf dem Balken ein schlechtes Gleichgewicht hatte und dazu neigte, ihre Schulter am Barren auszukugeln. Sie bemerkte, dass ihre Gelenke bei Bewegung knackten, und ließ sie oft absichtlich knacken, weil dies Spannung und Schmerzen zu lindern schien. Sie versuchte sich als Cheerleaderin und hatte Erfolg bei den Manövern, stellte aber fest, dass sie zu empfindlich auf Hitze reagierte. Die anderen Mädchen nannten sie „Tomatengesicht“, weil sie während der Übungen überhitzte, und sie ermüdete leicht und brauchte nach dem Training oft einen Tag Pause. Ihre Verdauungsprobleme machten sich durch häufige Verstopfung und Magenschmerzen bemerkbar, sie fühlte sich oft aufgebläht und brauchte Abführmittel. In der 6. Klasse fehlte sie wegen ihrer Darmprobleme und Gelenkschmerzen in der Schule und fühlte sich oft müde und schwindlig. Sie fühlte sich morgens nicht ausgeruht und hielt am Wochenende oft ein Nickerchen – eine Abkehr von ihrem früheren aktiven und enthusiastischen Lebensstil. Sie fühlte sich oft ängstlich, ohne dass sie dies auf einen bestimmten schulischen oder familiären Stress zurückführen konnte, und begann, morgens Kopfschmerzen zu haben, von denen sie annahm, dass sie auf ihren schlechten Schlaf zurückzuführen waren. Ihre Eltern brachten sie erneut zu ihrem Kinderarzt und erfuhren, dass es sich um den üblichen Schulstress handelte.
Als Eve 14 Jahre alt war, hatte sie eine schwere grippeähnliche Krankheit und fehlte einen Monat lang in der Schule, weil sie sehr müde war, Kopfschmerzen, Übelkeit, Erbrechen und Gewichtsverlust hatte. Sie litt unter furchtbarem Schwindel und Schmerzen in der Brust und hatte manchmal das Gefühl, ihr Herz würde pochen (Herzklopfen). Ihre Eltern holten den Rat eines anderen Kinderarztes ein, der Eve zu einem Kardiologen schickte, der glaubte, dass ihre Tachykardie auf Stress und Angst im Zusammenhang mit ihrer Krankheit zurückzuführen sei. Er verabreichte Eve Propranolol, einen Betablocker, um ihre Herzfrequenz zu verlangsamen, aber sie wurde noch müder und war nun die meiste Zeit des Tages bettlägerig. Ihre Gelenkschmerzen nahmen nach ihrer Erkrankung zu, und sie blieb fast den ganzen Tag im Bett, war traurig und wurde ängstlich, wenn ihre Eltern versuchten, sie nach draußen zu bringen oder mit ihr spazieren zu fahren. Sie brachten sie zurück zum zweiten Kinderarzt, der sie an die Psychiatrie überwies, und es wurde mit der Einnahme von Zoloft begonnen, um die Ängste und Depressionen zu lindern. Nach ein paar Tagen bekam Eve einen Ausschlag mit Nesselsucht und musste das Medikament absetzen. Sie litt weiterhin unter Schwindelgefühlen und wurde von zwei weiteren Fachärzten untersucht: einem Gastroenterologen, der eine Zöliakie vermutete, und einem Hals-Nasen-Ohren-Arzt, der Schwindel diagnostizierte und sie in die Neurologie überwies. Sie erzählte diesem Arzt von Kopfschmerzen, die von ihrem Hinterkopf ausgingen, und es wurde eine MRT-Untersuchung des Kopfes in Rückenlage durchgeführt, die normal ausfiel.
Die Anerkennung der wahrscheinlichen AAD-Diagnose durch ihren Hausarzt führte zu einer Reihe von Überweisungen an andere Fachärzte, darunter auch an die Neurologie, wo eine MRT-Untersuchung des Kopfes in aufrechter Position die Herniation einer Chiari-Verformung zeigte. Ein Kardiologe dokumentierte einen abnormalen Kipptisch-Test zusammen mit einer geringen Darmmotilität und wies nach, dass ihre Angstzustände und ihre Stressmüdigkeit auf Dysautonomie zurückzuführen waren. Eine Ernährungsstrategie mit Flüssigkeitszufuhr, Salz und hohem Proteingehalt sowie eine Therapie mit Betablockern und Antihistaminika führte zu einer Besserung ihrer Müdigkeit und ihrer Darmsymptome sowie zu einer Linderung ihrer Kopfschmerzen. Es wurde entschieden, dass die Chiari-Hernie von 4-5 mm nicht ausreichte, um eine Operation zu erfordern, und ihre psychiatrische Behandlung wurde als Behandlung sekundärer Symptome einer medizinischen Störung anerkannt. Einige ihrer Fachärzte, die der klinischen Diagnose skeptisch gegenüberstanden, wurden durch einen Gentest überzeugt, der eine Veränderung in einem Gen für die Alpha-1-Kette des Typ-V-Kollagens, abgekürzt COL5A1, nachwies, die bei Patienten mit EDS häufig vorkommt. Eve hatte die Mutation von ihrer Mutter geerbt, die mildere AAD-Symptome hatte und diese nun besser in den Griff bekam.
Im Laufe mehrerer Monate wurden Eves Gesundheit und Vitalität wiederhergestellt und sie „bekam ihr Leben zurück“, wie sie und ihre Eltern sagen würden. Sie hatte zwar immer noch gelegentlich Gelenkschmerzen, Angstzustände und Müdigkeit, aber „kluges Training“ zum Aufbau eines Muskelschutzes um ihre Gelenke herum, Ernährungsansätze gegen Dysautonomie und Biofeedback-Ansätze gegen Angstzustände waren lebensverändernd. Diese Behandlungen wurden von ihrem Hausarzt anerkannt, der nun in der Lage ist, die Betreuung von Eve und ihrer Mutter zu koordinieren und Ressourcen für das Verständnis des EDS und der zugrunde liegenden Genveränderungen bereitzustellen. Der Übergang zur nächsten Generation bestand darin, dass Eve über ihr 50-prozentiges Risiko, ihre COL5A-Mutation weiterzugeben, und über einige Risiken für frühe Wehen und postpartale Blutungen bei zukünftigen Schwangerschaften informiert werden konnte.
- Remvig L, Jensen DV, Ward RC (2007) Epidemiologie der allgemeinen Gelenkhypermobilität und Grundlage für die vorgeschlagenen Kriterien für das benigne Gelenkhypermobilitätssyndrom: Übersicht über die Literatur. J Rheumatol 34: 804-809.
- Steinman B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. In: Connective Tissue and its Heritable Disorders. B Steinman, PM Royce (Eds) Wiley-Liss: New York pp. 351-407.
- McKusick VA (1956) Heritable disorders of connective tissue. CV Mosby: St. Louis.
- Bloom L, Byers P, Francomano C, Tinkle B, Malfait F (2017) The International Consortium on the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175: 5-7.
- Gazit Y1, Nahir AM, Grahame R, Jacob G (2003) Dysautonomia in the joint hypermobility syndrome. Am J Med 115: 33-40.
- Pizzo PA1 (2013) Lessons in pain relief–a personal postgraduate experience. N Engl J Med 369: 1092-1093.
- Pyeritz RE (2008) Ein kleines Molekül für eine große Krankheit. N Engl J Med 358: 2829-2831.
- 8. Wyandt HE, Wilson GN, Tonk VS. Kapitel 11: Gen- und Genomsequenzierung: Interpretation der genetischen Variation auf Nukleotidebene. In: Human Chromosome Variation: Heteromorphism, Polymorphism, and Pathogenesis, Ed.2. Springer Nature, Singapore
- Mefford HC (2012) Diagnostic exome sequencing–are we there yet? N Engl J Med 367: 1951-1953.
- http://hypermobility.org/help-advice/hypermobility-syndromes/beighton-score/
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 12: 2989-3004.
- Simpson MR (2006) Benign joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc 106: 531-536.
- Kirk JA, Ansell BM, Bywaters EG (1967) The hypermobility syndrome. Muskuloskelettale Beschwerden im Zusammenhang mit generalisierter Gelenkhypermobilität. Ann Rheum Dis 26: 419-425.
- Wilson GN (2014) Presymptomatic and preimplantation genetic diagnosis: Neurologie, NextGenetics, und die nächste Generation. JAMA Neurol 71: 403-404.
- Castori M, Morlino S, Celletti C (2012) Management von Schmerzen und Müdigkeit beim Hypermobilitätssyndrom der Gelenke (auch bekannt als Ehlers-Danlos-Syndrom, Hypermobilitätstyp): Grundsätze und Vorschlag für einen multidisziplinären Ansatz. Am J Med Genet A 158: 2055-2070.
- Castori M, Morlino S, Celletti C (2013) Rewriting the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet A 161A: 2989-3004.
- Bathen T, Hangmann AB, Hoff M (2013) Multidisciplinary treatment of disability in Ehlers-Danlos syndrome hypermobility type/ hypermobility syndrome: a pilot study using a combination of physical and cognitivebehavioral therapy on 12 women. Am J Med Genet A 161A: 3005-3011.
- EDS (2018) Ehlers-Danlos Syndrome National Foundation.
- OMIM (2018) Online Mendelian Inheritance in Man.
- Wilson GN (2014) Exome analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am J Med Genet A 164A: 1209-1212.