Il ne faut pas s’étonner que, même dans les parties du génome où nous ne voyons pas manifestement un code « fonctionnel » (c’est-à-dire un code qui a été fixé par l’évolution à la suite d’un avantage sélectif quelconque), il y a un type de code, mais qui ne ressemble à rien de ce que nous avons considéré comme tel auparavant. Et s’il faisait quelque chose en trois dimensions en plus de la dimension unique du code ATGC ? Un article qui vient d’être publié dans BioEssays explore cette possibilité alléchante…
N’est-ce pas merveilleux d’avoir un problème vraiment perplexe à ronger, un qui génère des explications potentielles presque infinies. Que diriez-vous de « que fait tout cet ADN non codant dans les génomes ? » – ces 98,5% du matériel génétique humain qui ne produisent pas de protéines. Pour être honnête, le décryptage de l’ADN non codant progresse à grands pas grâce à l’identification de séquences transcrites en ARN qui modulent l’expression des gènes, peuvent être transmises de manière transgénérationnelle (épigénétique) ou définissent le programme d’expression génétique d’une cellule souche ou d’une cellule tissulaire spécifique. Des quantités massives de séquences répétées (vestiges d’anciens rétrovirus) ont été trouvées dans de nombreux génomes. Là encore, elles ne codent pas pour des protéines, mais il existe au moins des modèles crédibles de leur action en termes d’évolution (allant du parasitisme génomique à la symbiose, voire à l' »exploitation » par le génome hôte lui-même pour produire la diversité génétique sur laquelle fonctionne l’évolution) ; incidemment, certains ADN non codants fabriquent des ARN qui réduisent au silence ces séquences rétrovirales, et l’ingression rétrovirale dans les génomes aurait été la pression sélective pour l’évolution de l’interférence ARN (dite ARNi) ; les éléments répétitifs de divers types nommés et les répétitions en tandem abondent ; les introns (dont beaucoup contiennent les types de séquences non codantes susmentionnées) ont transpiré pour être cruciaux dans l’expression et la régulation des gènes, de manière plus frappante via l’épissage alternatif des segments codants qu’ils séparent.
Pour autant, il y a beaucoup de problèmes à ronger car, bien que nous comprenions de mieux en mieux la nature et l’origine d’une grande partie du génome non codant et que nous fassions des percées majeures dans sa « fonction » (définie ici comme un effet avantageux sélectionné par l’évolution sur l’organisme hôte), nous sommes loin de tout expliquer et, plus précisément, nous le regardons avec une lentille à très faible grossissement, pour ainsi dire. Les génomes viraux sont des exemples classiques où les gènes lus dans un sens pour produire une protéine donnée se superposent à un ou plusieurs gènes lus dans le sens opposé (c’est-à-dire à partir du brin complémentaire de l’ADN) pour produire des protéines différentes. C’est un peu comme faire des messages simples avec des mots en paires inversées (ce qu’on appelle un emordnilap). Par exemple : REEDSTOPSFLOW, qui, par un dispositif de lecture imaginaire, pourrait être divisé en REED STOPS FLOW. Lu à l’envers, cela donnerait WOLF SPOTS DEER.
Maintenant, s’il est avantageux pour l’évolution que deux messages soient codés de manière aussi économique – comme c’est le cas dans les génomes viraux, qui ont tendance à évoluer vers une complexité minimale en termes de contenu d’information, réduisant ainsi les ressources nécessaires à la reproduction – alors les messages eux-mêmes évoluent avec un haut degré de contrainte. Qu’est-ce que cela signifie ? Eh bien, nous pourrions formuler le message de notre exemple initial comme suit : RUSH-STEM IMPEDES CURRENT, qui contiendrait la même information essentielle que REED STOPS FLOW. Cependant, ce message, s’il est lu à l’envers (ou même dans le même sens, mais dans des parties différentes), ne contient rien de plus qui soit particulièrement significatif. La seule façon de transmettre simultanément les deux informations contenues dans les messages originaux est probablement la formulation même de REEDSTOPSFLOW : c’est un système très contraint ! En effet, si l’on étudiait suffisamment d’exemples de phrases inversées en anglais, on constaterait qu’elles sont, dans l’ensemble, composées de mots plutôt courts, et que les séquences manquent de certaines unités de langage comme les articles (the, a) ; si l’on regardait de plus près, on pourrait même détecter une représentation supérieure à la moyenne de certaines lettres de l’alphabet dans ces messages. Nous verrions cela comme des biais dans l’utilisation des mots et des lettres qui nous permettraient, a priori, d’avoir un coup de couteau pour identifier de tels éléments d’information à « double fonction ».
Revenons maintenant aux « lettres », aux « mots » et aux « informations » codées dans les génomes. Pour que deux informations distinctes soient codées dans le même morceau de séquence génétique, nous nous attendrions, de la même manière, à ce que les contraintes se manifestent par des biais d’utilisation des mots et des lettres – analogies, respectivement, avec les séquences d’acides aminés constituant les protéines, et leur code à trois lettres. Ainsi, une séquence d’ADN peut coder pour une protéine et, en outre, pour autre chose. Ce « quelque chose d’autre », selon Giorgio Bernardi, est l’information qui dirige l’emballage de l’énorme longueur d’ADN dans une cellule dans le noyau relativement minuscule. Il s’agit principalement du code qui guide la liaison des protéines d’emballage de l’ADN, les histones. Bernardi appelle cela le « code génomique » – un code structurel qui définit la forme et la compaction de l’ADN dans la forme hautement condensée connue sous le nom de « chromatine ».
Mais n’avons-nous pas commencé par une explication pour l’ADN non codant, et non pour les séquences codant pour les protéines ? Oui, et dans les longs tronçons d’ADN non codant, nous voyons des informations qui dépassent les simples répétitions, les répétitions en tandem et les restes d’anciens rétrovirus : il existe un type de code au niveau de la préférence pour la paire GC des bases chimiques de l’ADN par rapport à l’AT. Comme l’explique Bernardi, qui synthétise ses travaux révolutionnaires et ceux d’autres chercheurs, dans les séquences de base du génome eucaryote, la teneur en GC dans les unités organisationnelles structurelles du génome appelées « isochores » a augmenté au cours de la transition évolutive entre les organismes dits à sang froid et ceux à sang chaud. Et, de manière fascinante, ce biais de séquence se superpose à des séquences qui sont beaucoup plus contraintes dans leur fonction : ce sont les séquences codant pour les protéines mêmes mentionnées précédemment, et elles – plus que les séquences non codantes intermédiaires – sont l’indice du « code génomique ».
Les séquences codant pour les protéines sont également emballées et condensées dans le noyau – en particulier lorsqu’elles ne sont pas « utilisées » (c’est-à-dire, en train d’être transcrites, puis traduites en protéines) – mais elles contiennent aussi des informations relativement constantes sur les identités précises des acides aminés, sinon elles ne parviendraient pas à coder correctement les protéines : l’évolution agirait sur ces mutations de manière très négative, ce qui les rendrait extrêmement improbables pour persister et être visibles pour nous. Mais le code des acides aminés dans l’ADN comporte un petit « piège » qui a évolué dans les organismes unicellulaires les plus simples (bactéries et archées) il y a des milliards d’années : le code est partiellement redondant. Par exemple, l’acide aminé Thréonine peut être codé dans l’ADN eucaryote de pas moins de quatre façons : ACT, ACC, ACA ou ACG. La troisième lettre est variable et donc « disponible » pour le codage d’informations supplémentaires. C’est exactement ce qui se passe pour produire le « code génomique », créant dans ce cas un biais pour les formes ACC et ACG chez les organismes à sang chaud. Par conséquent, la forte contrainte qui pèse sur ce « code » supplémentaire – que l’on observe également dans des parties du génome qui ne sont pas soumises à une telle contrainte comme les séquences codant pour les protéines – est imposée par l’empaquetage des séquences codant pour les protéines qui incarnent deux ensembles d’informations simultanément. Ceci est analogue à notre exemple de la séquence à double information fortement contrainte REEDSTOPSFLOW.
Important, cependant, la contrainte n’est pas aussi stricte que dans notre exemple en langue anglaise en raison de la redondance de la troisième position du code triplet pour les acides aminés : une meilleure analogie serait SHE*ATE*STU* où l’astérisque représente une lettre variable qui ne fait aucune différence pour la machine qui lit la composante à trois lettres du message à quatre lettres. On pourrait ensuite imaginer un deuxième niveau d’information formé en ajoutant un « D » à ces points d’astérisque, pour obtenir SHEDATEDSTUD (SHE DATED STUD). Imaginez ensuite une deuxième machine à lire qui recherche des phrases significatives de « nature sensible » contenant une concentration de D supérieure à la moyenne. Cette machine à lire est accompagnée d’une machine à plier qui place une sorte de cheville à chaque D, pliant le message de 120 degrés dans un plan. un point où le message devrait être plié de 120 degrés dans le même plan, nous obtiendrions une version plus compacte, triangulaire. Dans les génomes eucaryotes, le biais de la séquence GC proposé comme responsable de la condensation structurelle s’étend aux séquences non codantes, dont certaines ont des activités identifiées, bien que moins contraintes dans leur séquence que l’ADN codant pour les protéines. Là, il dirige leur condensation via les nucléosomes contenant des histones pour former la chromatine.
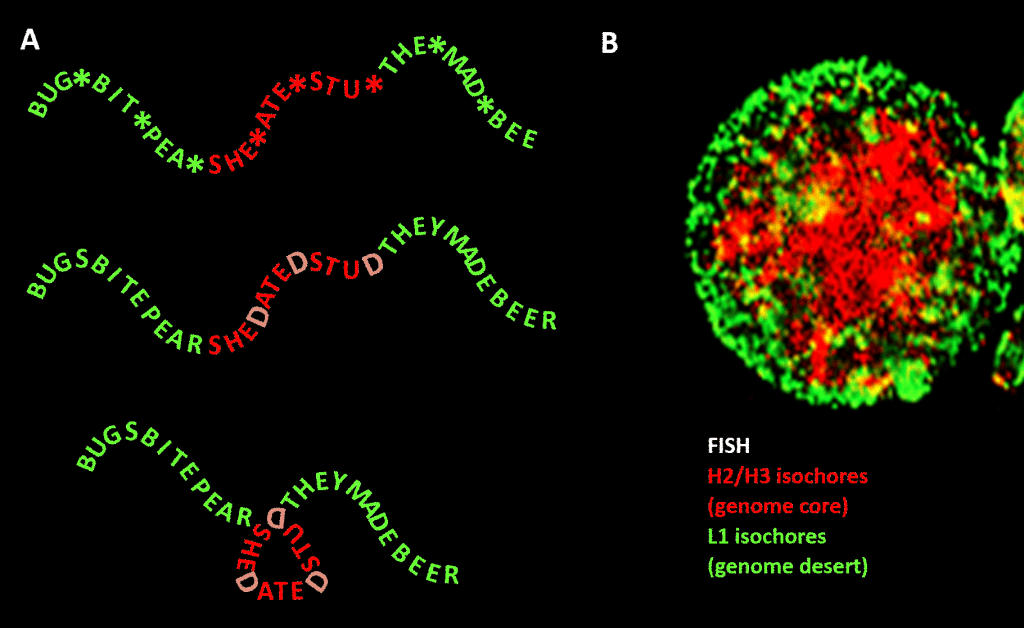
Figure. Analogie entre la condensation d’un message à base de mots et la condensation de l’ADN génomique dans le noyau cellulaire. Panneau A : information dans l’information, une séquence de mots avec un quatrième espace variable qui, lorsqu’il est rempli de lettres particulières, génère un autre message. Un message est lu par une machine de lecture à trois lettres ; l’autre par une machine de lecture qui peut interpréter des informations s’étendant jusqu’à la quatrième position variable de la séquence. Le second lecteur reconnaît les informations « sensibles » qui doivent être dissimulées, et aux endroits où un « D » apparaît en 4ème position, il plie la chaîne de mots, comprimant ainsi la partie « sensible » et la mettant hors de vue. Il s’agit d’une analogie avec le principe de la compression génomique en 3D par la chromatine, comme le montre le panneau B : une image de fluorescence (par hybridation in situ en fluorescence – FISH) du noyau cellulaire. Les isochores H2/H3, dont le contenu en GC a augmenté au cours de l’évolution des vertébrés à sang froid vers les vertébrés à sang chaud, sont comprimés dans un noyau de chromatine, laissant les isochores L1 (avec un contenu en GC plus faible) à la périphérie dans un état moins condensé. Selon Bernardi, le « code génomique » incarné dans les segments à forte teneur en GC du génome est lu par la machinerie de positionnement des nucléosomes de la cellule et interprété comme une séquence devant être fortement comprimée dans l’euchromatine. Remerciements : Panel A : concept et production de figures : Andrew Moore ; Panneau B : Un schéma FISH d’isochores H2/H3 et L1 provenant d’un lymphocyte induit par PHA – avec l’aimable autorisation de S. Saccone – tel que reproduit dans la Réf…].
Ces régions de l’ADN peuvent alors être considérées comme des éléments structurellement importants pour former la forme et la séparation correctes des séquences codantes condensées dans le génome, indépendamment de toute autre fonction possible de ces séquences non codantes : en substance, il s’agirait d’une » explication » de la persistance dans les génomes de séquences auxquelles aucune » fonction » (en termes d’activité sélectionnée par l’évolution), ne peut être attribuée (ou, du moins, aucune fonction substantielle).
Une dernière analogie – cette fois beaucoup plus étroitement liée – pourrait être les séquences d’acides très aminés dans les grandes protéines, qui font une variété de torsions, de tours, de plis, etc. Nous pouvons nous émerveiller devant des structures aussi compliquées et nous demander « mais ont-elles besoin d’être aussi compliquées pour leur fonction ? ». Eh bien, peut-être que oui, afin de condenser et de positionner les parties de la protéine dans l’orientation et à l’endroit exacts qui génèrent la structure tridimensionnelle qui a été sélectionnée avec succès par l’évolution. Mais en sachant que le « code génomique » chevauche les séquences codantes des protéines, nous pourrions même commencer à soupçonner qu’une autre pression sélective est également à l’œuvre…
Andrew Moore, Ph.D.
Éditeur en chef, BioEssays
.