Non dovrebbe sorprenderci che anche in parti del genoma dove non vediamo ovviamente un codice “funzionale” (cioè, uno che è stato fissato evolutivamente come risultato di qualche vantaggio selettivo), c’è un tipo di codice, ma non come qualcosa che abbiamo precedentemente considerato come tale. E se facesse qualcosa in tre dimensioni, oltre all’unica dimensione del codice ATGC? Un articolo appena pubblicato su BioEssays esplora questa allettante possibilità…
Non è meraviglioso avere un problema davvero sconcertante su cui rosicchiare, uno che genera spiegazioni potenziali quasi infinite. Che ne dite di “cosa ci fa tutto quel DNA non codificante nei genomi?” – quel 98,5% del materiale genetico umano che non produce proteine. Ad essere onesti, la decifrazione del DNA non codificante sta facendo grandi passi avanti attraverso l’identificazione di sequenze che sono trascritte in RNA che modulano l’espressione genica, possono essere trasmesse transgenerazionalmente (epigenetica) o impostare il programma di espressione genica di una cellula staminale o di una cellula specifica del tessuto. Quantità massicce di sequenze ripetute (resti di antichi retrovirus) sono state trovate in molti genomi, e di nuovo, queste non codificano per le proteine, ma almeno ci sono modelli credibili per quello che stanno facendo in termini evolutivi (che vanno dal parassitismo genomico alla simbiosi e persino allo “sfruttamento” da parte del genoma stesso ospite per produrre la diversità genetica su cui lavora l’evoluzione); per inciso, alcuni DNA non codificanti producono RNA che mettono a tacere queste sequenze retrovirali, e si ritiene che l’ingressione retrovirale nei genomi sia stata la pressione selettiva per l’evoluzione dell’interferenza dell’RNA (il cosiddetto RNAi); abbondano gli elementi ripetitivi di vari tipi nominati e le ripetizioni tandem; gli introni (molti dei quali contengono i suddetti tipi di sequenze non codificanti) si sono rivelati cruciali nell’espressione e regolazione genica, soprattutto attraverso lo splicing alternativo dei segmenti codificanti che essi separano.
Ancora, ci sono molti problemi da risolvere, perché anche se stiamo comprendendo sempre di più la natura e l’origine di gran parte del genoma non codificante e stiamo facendo grandi scoperte sulla sua “funzione” (definita qui come effetto vantaggioso selezionato evolutivamente sull’organismo ospite), siamo lontani dallo spiegarlo tutto, e – più precisamente – lo stiamo guardando con una lente di ingrandimento molto bassa, per così dire. Una delle cose intriganti delle sequenze di DNA è che una singola sequenza può “codificare” più di un’informazione a seconda di ciò che la “legge” e in quale direzione – i genomi virali sono esempi classici in cui i geni letti in una direzione per produrre una data proteina si sovrappongono a uno o più geni letti nella direzione opposta (cioè, dal filamento complementare di DNA) per produrre proteine diverse. È un po’ come fare semplici messaggi con parole a coppia inversa (un cosiddetto emordnilap). Per esempio: REEDSTOPSFLOW, che, con un immaginario dispositivo di lettura, potrebbe essere diviso in REED STOPS FLOW. Letto al contrario, darebbe WOLF SPOTS DEER.
Ora, se è un vantaggio evolutivo che due messaggi siano codificati in modo così economico – come nel caso dei genomi virali, che tendono ad evolvere verso una complessità minima in termini di contenuto informativo, riducendo così le risorse necessarie alla riproduzione – allora i messaggi stessi evolvono con un alto grado di costrizione. Che cosa significa questo? Bene, potremmo formulare il nostro messaggio originale di esempio come RUSH-STEM IMPEDES CURRENT, che racchiuderebbe la stessa informazione essenziale di REED STOPS FLOW. Tuttavia, quel messaggio, se letto al contrario (o anche nello stesso senso, ma in pezzi diversi) non codifica nulla di aggiuntivo che sia particolarmente significativo. Probabilmente l’unico modo di trasmettere simultaneamente entrambe le informazioni nei messaggi originali è proprio la dicitura REEDSTOPSFLOW: è un sistema altamente vincolato! Infatti, se studiassimo abbastanza esempi di frasi a coppie inverse in inglese, vedremmo che sono, nel complesso, costituite da parole piuttosto brevi, e le sequenze mancano di alcune unità linguistiche come gli articoli (the, a); se guardassimo più da vicino, potremmo persino rilevare una rappresentazione maggiore della media di alcune lettere dell’alfabeto in tali messaggi. Vedremmo queste come distorsioni nell’uso delle parole e delle lettere che ci permetterebbero, a priori, di avere un tentativo di identificare tali informazioni “a doppia funzione”.
Ora torniamo alle “lettere”, “parole” e “informazioni” codificate nei genomi. Per due informazioni distinte da codificare nello stesso pezzo di sequenza genetica, ci aspetteremmo, allo stesso modo, che i vincoli si manifestino in distorsioni nell’uso delle parole e delle lettere – le analogie, rispettivamente, per le sequenze di amminoacidi che costituiscono le proteine e il loro codice a tre lettere. Quindi una sequenza di DNA può codificare una proteina e, in aggiunta, qualcos’altro. Questo “qualcos’altro”, secondo Giorgio Bernardi, è l’informazione che dirige il confezionamento dell’enorme lunghezza del DNA di una cellula nel nucleo relativamente piccolo. Principalmente è il codice che guida il legame delle proteine di impacchettamento del DNA note come istoni. Bernardi si riferisce a questo come al “codice genomico” – un codice strutturale che definisce la forma e la compattazione del DNA nella forma altamente condensata nota come “cromatina”.
Ma non abbiamo iniziato con una spiegazione del DNA non codificante, non delle sequenze codificanti le proteine? Sì, e nei lunghi tratti di DNA non codificante vediamo informazioni in eccesso rispetto a semplici ripetizioni, ripetizioni in tandem e resti di antichi retrovirus: c’è un tipo di codice a livello di preferenza per la coppia GC di basi chimiche del DNA rispetto all’AT. Come Bernardi ha esaminato, sintetizzando il suo lavoro rivoluzionario e quello di altri, nelle sequenze centrali del genoma eucariotico, il contenuto di GC nelle unità organizzative strutturali del genoma chiamate “isocore” è aumentato durante la transizione evolutiva tra i cosiddetti organismi a sangue freddo e quelli a sangue caldo. E, cosa affascinante, questa polarizzazione delle sequenze si sovrappone a sequenze che sono molto più vincolate nella funzione: queste sono proprio le sequenze codificanti le proteine menzionate prima, e sono – più delle sequenze non codificanti che intervengono – l’indizio del “codice genomico”.
Le sequenze codificanti le proteine sono anche imballate e condensate nel nucleo – soprattutto quando non sono “in uso” (cioè essere trascritte e poi tradotte in proteine) – ma contengono anche informazioni relativamente costanti sulle identità precise degli amminoacidi, altrimenti non riuscirebbero a codificare correttamente le proteine: l’evoluzione agirebbe su tali mutazioni in modo altamente negativo, rendendone estremamente improbabile la persistenza e la visibilità per noi. Ma il codice degli aminoacidi nel DNA ha un piccolo “tranello” che si è evoluto nel più semplice degli organismi unicellulari (batteri e archaea) miliardi di anni fa: il codice è in parte ridondante. Per esempio, l’aminoacido treonina può essere codificato nel DNA eucariotico in non meno di quattro modi: ACT, ACC, ACA o ACG. La terza lettera è variabile e quindi “disponibile” per la codifica di informazioni extra. Questo è esattamente ciò che accade per produrre il “codice genomico”, in questo caso creando una preferenza per le forme ACC e ACG negli organismi a sangue caldo. Quindi, l’alto vincolo di questo “codice” addizionale -che si vede anche in parti del genoma che non sono sotto tale vincolo come sequenze codificanti le proteine- è imposto dal confezionamento di sequenze codificanti le proteine che incarnano due serie di informazioni simultaneamente. Questo è analogo al nostro esempio della sequenza di doppia informazione altamente vincolata REEDSTOPSFLOW.
Importante, tuttavia, il vincolo non è così stretto come nel nostro esempio di lingua inglese a causa della ridondanza della terza posizione della tripletta di codice per gli aminoacidi: una migliore analogia sarebbe SHE*ATE*STU* dove l’asterisco sta per una lettera variabile che non fa alcuna differenza per la macchina che legge la componente a tre lettere del messaggio a quattro lettere. Si potrebbe poi immaginare un secondo livello di informazione formato dall’aggiunta di “D” in questi punti di asterisco, per fare SHEDATEDSTUD (SHE DATED STUD). Poi immaginate una seconda macchina di lettura che cerca frasi significative di “natura sensibile” contenenti una concentrazione di D superiore alla media. Questa macchina di lettura porta con sé una macchina piegatrice che mette una specie di piolo ad ogni D, piegando il messaggio di 120 gradi in un piano. un punto in cui il messaggio dovrebbe essere piegato di 120 gradi nello stesso piano, ci ritroveremmo con una versione più compatta, triangolare. Nei genomi eucarioti, il bias di sequenza GC proposto per essere responsabile della condensazione strutturale si estende nelle sequenze non codificanti, alcune delle quali hanno attività identificate, anche se meno vincolate nella sequenza rispetto al DNA codificante le proteine. Lì dirige la loro condensazione attraverso nucleosomi contenenti istoni per formare la cromatina.
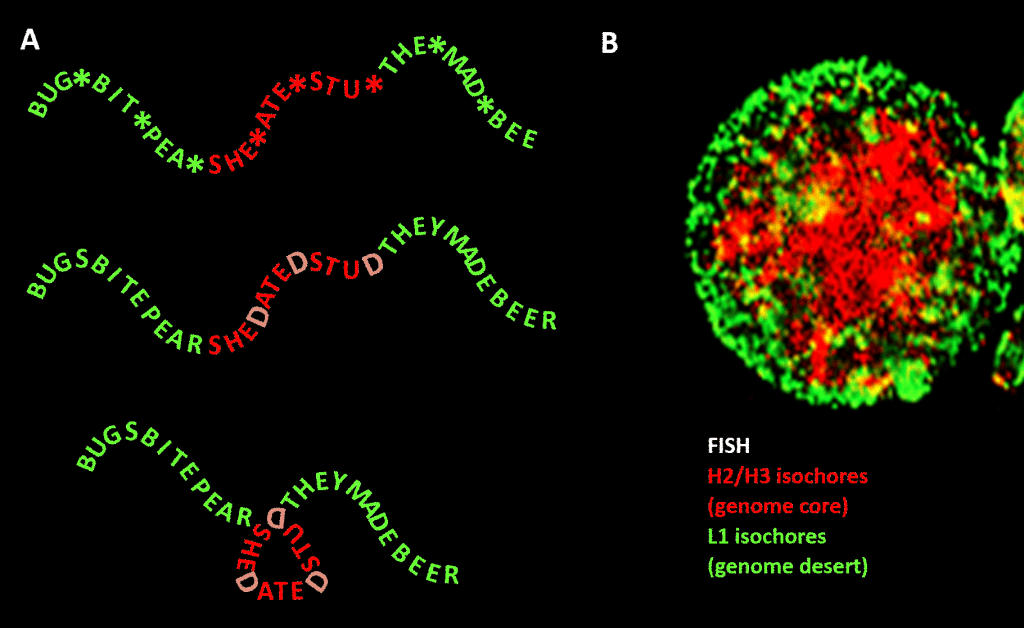
Figura. Analogia tra la condensazione di un messaggio basato su parole e la condensazione del DNA genomico nel nucleo della cellula. Pannello A: Informazione nell’informazione, una sequenza di parole con un quarto spazio variabile che, se riempito con particolari lettere, genera un ulteriore messaggio. Un messaggio viene letto da una macchina di lettura a tre lettere; l’altro da una macchina di lettura che può interpretare l’informazione che si estende fino alla quarta posizione variabile della sequenza. Il secondo lettore riconosce l’informazione “sensibile” che deve essere nascosta, e nei punti in cui appare una “D” in 4a posizione, piega la stringa di parole, comprimendo così la parte “sensibile” e togliendola alla vista. Questa è un’analogia per il principio della compressione 3D genomica attraverso la cromatina, come illustrato nel pannello B: un’immagine di fluorescenza (tramite l’ibridazione in situ di fluorescenza – FISH) del nucleo della cellula. Gli isocori H2/H3, che sono aumentati in contenuto di GC durante l’evoluzione dai vertebrati a sangue freddo a quelli a sangue caldo, sono compressi in un nucleo di cromatina, lasciando gli isocori L1 (con contenuto di GC inferiore) alla periferia in uno stato meno condensato. Il “codice genomico” incarnato nei tratti ad alto GC del genoma è, secondo Bernardi, letto dal macchinario di posizionamento dei nucleosomi della cellula e interpretato come sequenza da comprimere altamente nell’eucromatina. Ringraziamenti: Pannello A: concetto e produzione della figura: Andrew Moore; Pannello B: un modello FISH di isocore H2/H3 e L1 da un linfocita indotto da PHA – per gentile concessione di S. Saccone – come riprodotto in Rif.]
Queste regioni di DNA possono quindi essere considerate come elementi strutturalmente importanti nel formare la corretta forma e separazione delle sequenze codificanti condensate nel genoma, indipendentemente da qualsiasi altra possibile funzione che quelle sequenze non codificanti hanno: in sostanza, questa sarebbe una “spiegazione” per la persistenza nei genomi di sequenze a cui nessuna “funzione” (in termini di attività evolutivamente selezionata), può essere attribuita (o, almeno, nessuna funzione sostanziale).
Un’ultima analogia – questa volta molto più strettamente correlata – potrebbe essere quella delle sequenze di aminoacidi nelle grandi proteine, che fanno una varietà di torsioni, giri, pieghe ecc. Potremmo meravigliarci di tali strutture complicate e chiedere “ma hanno bisogno di essere così complicate per la loro funzione? Beh, forse lo fanno per condensare e posizionare parti della proteina nell’esatto orientamento e luogo che genera la struttura tridimensionale che è stata selezionata con successo dall’evoluzione. Ma sapendo che il “codice genomico” si sovrappone alle sequenze codificanti delle proteine, potremmo anche iniziare a sospettare che ci sia anche un’altra pressione selettiva all’opera…
Andrew Moore, Ph.D.
Editor-in-Chief, BioEssays