De sex viktigaste elektrofila aromatiska substitutionsreaktionerna
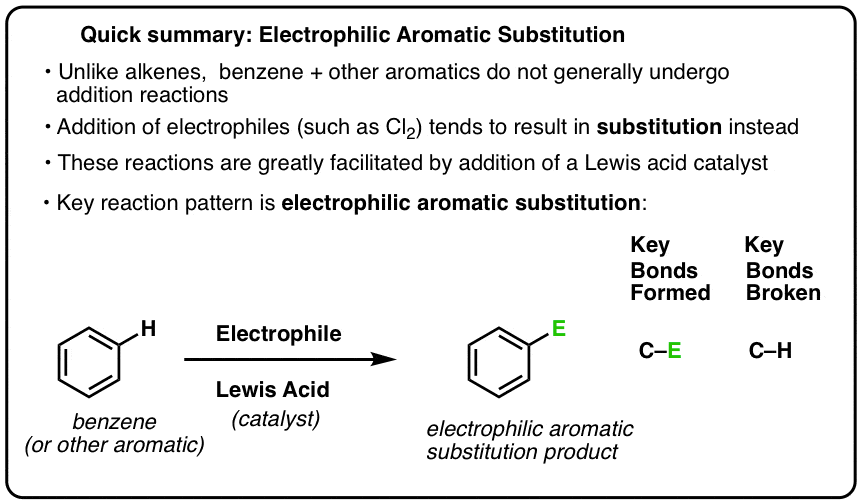
En nyckelreaktion för aromatiska föreningar är elektrofil aromatisk substitution, där en C-H-bindning bryts och en ny C-E-bindning bildas (E är en elektrofil atom, t.ex. Cl, Br, N…).
Innehållsförteckning
- Alkener ger ”additions”-produkter vid reaktion med elektrofiler. Så hur jämför sig bensen?
- Elektrofil aromatisk substitution
- Lewis-syror påskyndar hastigheten av elektrofila aromatiska substitutionsreaktioner
- Nyckelmönstret för sex viktiga elektrofila aromatiska substitutionsreaktioner
- Aromatisk klorering och brominering
- Aromatisk nitrering och sulfonering
- Friedel-Crafts alkylering och acylering
- Sammanfattning: De sex viktigaste elektrofila aromatiska substitutionsreaktionerna
- Quiz yourself!
- Alkener ger ”additionsprodukter” vid reaktion med elektrofiler. Så hur jämför bensen?
- Elektrofil aromatisk substitution
- Lewis-syror påskyndar hastigheten av elektrofila aromatiska substitutionsreaktioner
- Nyckelmönstret för sex viktiga elektrofila aromatiska substitutionsreaktioner
- Klorinering och brominering av aromatiska molekyler
- Nitration och sulfonering av aromatiska molekyler
- Friedel-Crafts alkylering och acylering
- Sammanfattning: De sex viktigaste elektrofila aromatiska substitutionsreaktionerna
- Testa dig själv!
Alkener ger ”additionsprodukter” vid reaktion med elektrofiler. Så hur jämför bensen?
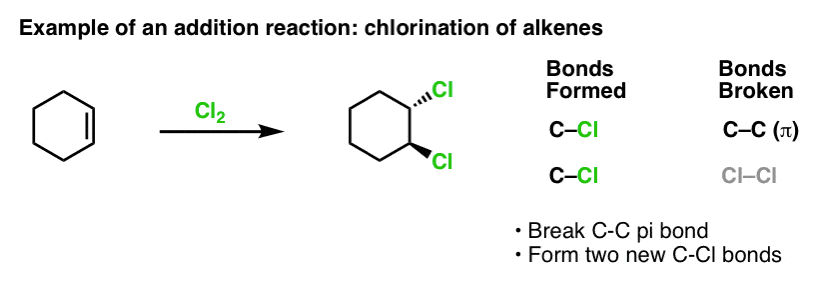
När vi behandlade reaktionerna hos alkener för ett tag sedan – många reaktioner! – såg vi att de allra flesta föll in i den klass av reaktioner som vi kallar additionsreaktioner. Det är när vi bryter en (relativt svag) C-C (pi)-bindning och bildar två nya enkelbindningar till kol. Klorering av alkener med Cl2 är ett klassiskt exempel:
När vi ändå är inne på ämnet bensen är det naturligt att fråga sig hur väl pi-bindningarna i aromatiska system (som bensen) står sig i reaktivitet i förhållande till pi-bindningarna i alkener, och i förlängningen hur reaktionerna hos aromatiska föreningar står sig i förhållande till reaktionerna hos alkener.
Vi har sett att bensen har en ovanlig stabilitet (36 kcal/mol resonansenergi) i förhållande till vad vi skulle förvänta oss för teoretisk ”cyklohexatrien”, vilket säkerligen skulle leda oss till att förutsäga att pi-bindningarna i aromatiska molekyler kommer att vara mindre reaktiva, i förhållande till alkener.
Du kanske också minns att bensen i sig självt är ovanligt svårt att hydrogenera. Väte (H2) kan fås att lägga sig över de flesta alkener i närvaro av en katalytisk mängd finfördelat palladium på kol (Pd/C), men man måste verkligen ta fram burken med Aldrich Brand Whup-Ass® (dvs. höga temperaturer, höga tryck av H2 , förlängda reaktionstider ) för att lyckas addera H2 till bensen, i förhållande till ”typiska” alkener.
Med tanke på detta, hur kan vi förvänta oss att elektrofiler som Cl2 eller Br2 reagerar med aromatiska föreningar som bensen?
Elektrofil aromatisk substitution
Vår första gissning skulle kunna vara att bensen reagerar med Cl2 för att ge en ”additions”-produkt som den som visas nedan (om än långsammare än med en ”normal” alken):
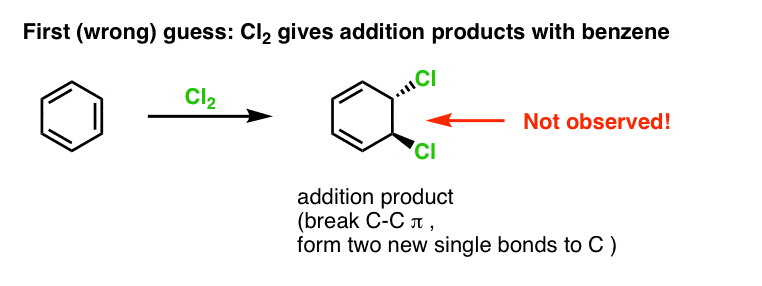
Det är faktiskt inte den produkt som vi observerar!
Om man istället behandlar bensen med Cl2 får man så småningom (och mycket långsamt) följande produkt: klorbensen.
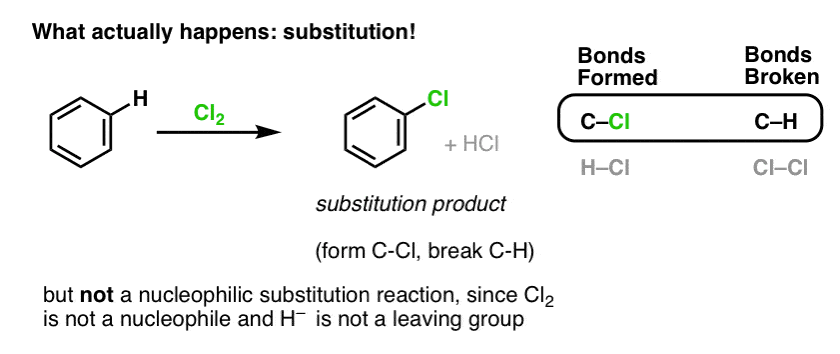
Vilka bindningar bildades och vilka bindningar bröts i denna reaktion?
Vi bildade C-Cl och bröt C-H . Eftersom pi-bindningarna alla är intakta är detta inte en additionsreaktion. Istället är detta alltså en typ av substitutionsreaktion, där vi bildar och bryter en bindning på ett enda kol.
Vi har sett ett exempel på substitutionsreaktioner tidigare, men det var nukleofila substitutioner, där en nukleofil (t.ex. RS- ) läggs till en alkylhalogenidelektrofil (t.ex. R-Br), som förskjuter en avgående grupp (Br- här) och bildar en C-Nuc-bindning (C-S i det här fallet) och bryter en C-LG-bindning (C-Br här).
Är reaktionen av Cl2 med bensen likaså en nukleofil substitutionsreaktion? Nej!
Cl2 är en extremt dålig nukleofil och reagerar som elektrongivare endast med starka Lewis-syror (t.ex. AlCl3, nedan). När det kombineras med även en relativt mild nukleofil, som pi-bindningen i en alken, beter det sig som en elektrofil (elektronacceptor), vilket vi såg i dess reaktion med alkener.
Cl2:s reaktion med bensen kallas därför för en elektrofil aromatisk substitution (förkortat EAS):
- Elektrofil, eftersom vi lägger till en elektronfattig art (elektrofil),
- till en aromatisk förening (bensen);
- substitution, eftersom vi bryter C-H och bildar C-E, där E är vår elektrofil (Cl i detta fall).
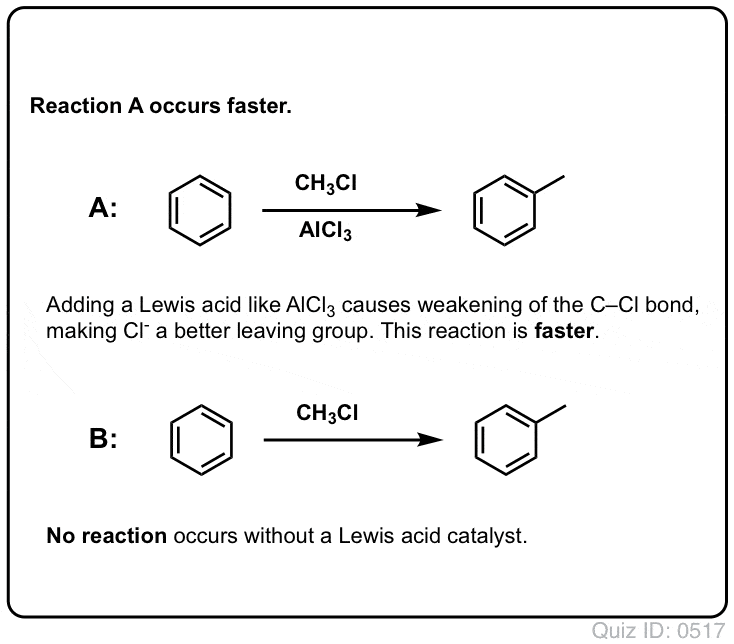
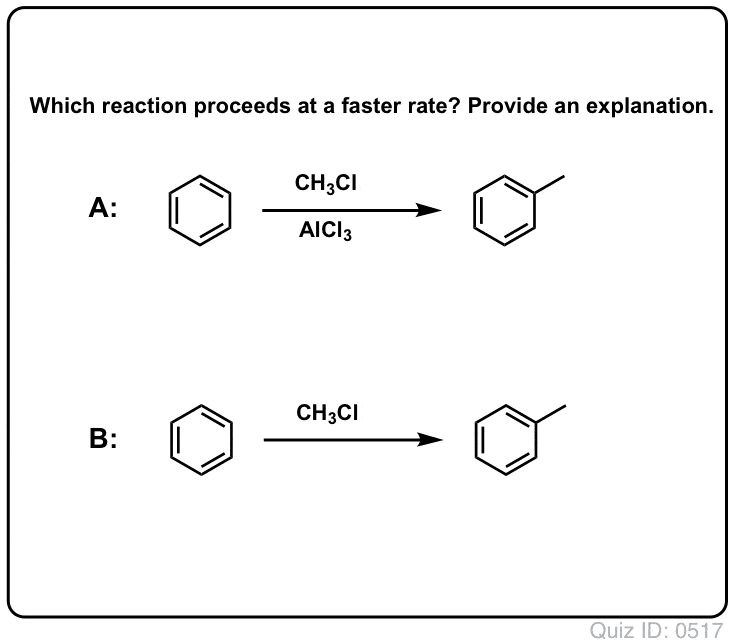
Lewis-syror påskyndar hastigheten av elektrofila aromatiska substitutionsreaktioner
Reaktionen av Cl2 med bensen är snabbare än tånageltillväxten, men inte särskilt mycket (Den är dock mycket snabbare med mer elektronrika aromater som toluen och fenol). Så istället för att sitta i labbet i veckor och vänta på att en reaktion ska slutföras kan vi tillsätta en reagens som ”soppar upp” reaktiviteten hos Cl2 för att göra den till en ännu bättre elektrofil: en katalysator, med andra ord.
Tillägg av en bra Lewis-syra som AlCl3 eller FeCl3 gör susen. . uppdatering: se inlägg om mekanism
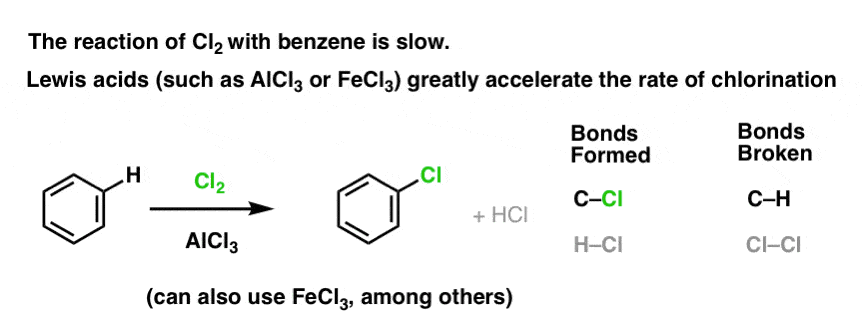
Hur fungerar detta?
Från vår serie om alkoholer minns du kanske att alkoholer (R-OH) kan övertalas att delta i substitutions- och eliminationsreaktioner om en stark syra tillsätts ( bildar R-OH2+ ). Det beror på att den konjugerade syran alltid är en bättre avgångsgrupp ; H2O är en svagare bas, och därmed en mycket bättre avgångsgrupp, än HO- . Etanol själv kommer aldrig att reagera med NaCl för att ge etylklorid, eftersom den resulterande avgångsgruppen HO- är en alltför stark bas i förhållande till Cl- för att reaktionen ska kunna fortgå i någon utsträckning. Men om vi omvandlar alkoholen till dess konjugerade syra R-OH2 + med en stark syra, t.ex. HCl, kan reaktionen fortsätta, eftersom Cl- tränger undan den mycket svagare basen H2O.
Det kan vara till hjälp att tänka sig att H+ försvagar C-O-bindningen och därmed gör kolet som är knutet till den till en bättre elektrofil.
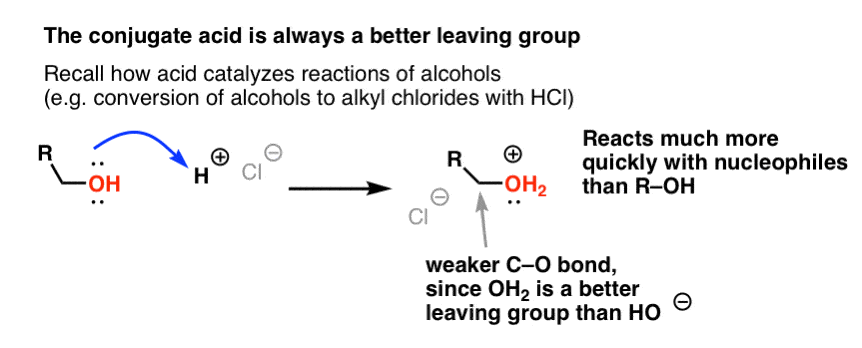
Precis som syra ”primar” det kol som är knutet till OH-gruppen för en efterföljande reaktion genom att omvandla hydroxylgruppen till dess konjugerade syra, utför Lewis-syror på liknande sätt samma magi på Cl2 (och för den delen kommer alla elektrofila aromatiska substitutionsreaktioner som vi kommer att behandla att innefatta någon form av syrakatalys).
I fallet med klorering tar Lewis-syran (AlCl3, nedan) emot ett par elektroner från Cl2. Detta försvagar Cl-Cl-bindningen och gör den till en ännu bättre nukleofil. Attack av en nukleofil vid det distala Cl frigör inte Cl- , utan den ännu svagare basen (och därmed bättre avgångsgrupp) AlCl4(-).
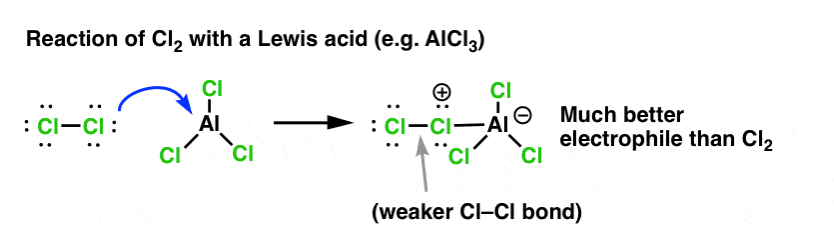
Tillägg av en Lewis-syra (som, kom ihåg, inkluderar Brønsted-syror som H2SO4 ) är en gemensam nämnare för de sex viktiga elektrofila aromatiska substitutioner som i allmänhet behandlas i introduktionskursen i organisk kemi.
Nyckelmönstret för sex viktiga elektrofila aromatiska substitutionsreaktioner
I det här avsnittet kommer vi att presentera dessa sex viktiga elektrofila aromatiska substitutionsreaktioner, som grupperar sig fint i tre par:
- klorering och bromering
- nitrering och sulfonylering
- Friedel-Crafts alkylering och Friedel-Crafts acylering
Vi ska inte gå in på mekanismerna ännu. Poängen här är bara att följa vilka bindningar som bildas och bryts, så att du ser nyckelmönstret.
Här är det:
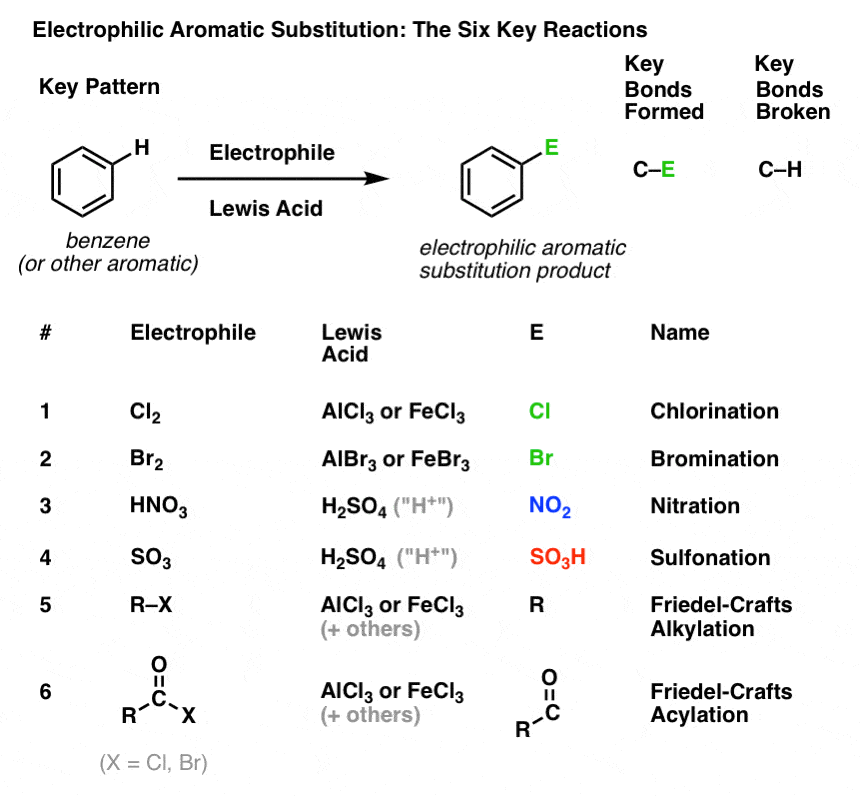
Klorinering och brominering av aromatiska molekyler
Vi har sett klorinering (ovan); brominering är mycket liknande. Eftersom Br2 i sig själv inte är en tillräckligt stark elektrofil för att reagera med bensen med en rimlig hastighet, använder vi en Lewis-syra som AlBr3 eller FeBr3 för att påskynda reaktionen. .
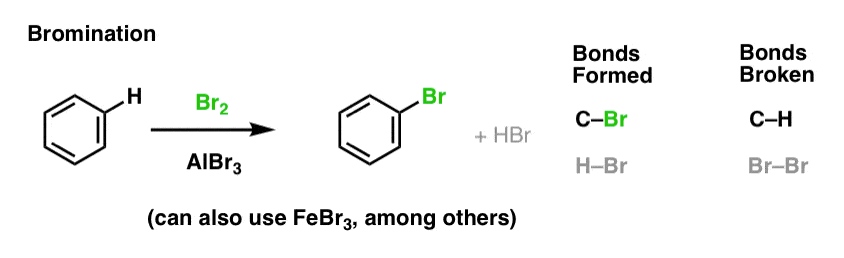
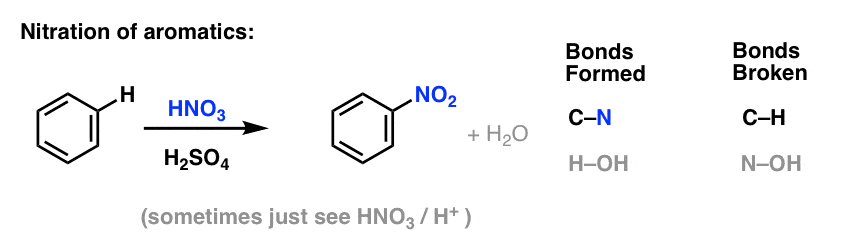
Nitration och sulfonering av aromatiska molekyler
Nitration (ersättning av H med en NO2-grupp) är inte en reaktion som vi såg i vårt avsnitt om alkener, men det är en populär reaktion med aromatiska molekyler som bensen. Detta är den reaktion genom vilken metylbensen (toluen) omvandlas till 2,4,6-trinitrotoluen. Du är troligen mer bekant med denna molekyl som det högexplosiva TNT.
Nitration är ersättningen av H med NO2, med salpetersyra (HNO3) som NO2-källa och svavelsyra (H2SO4) som Lewis-syra:
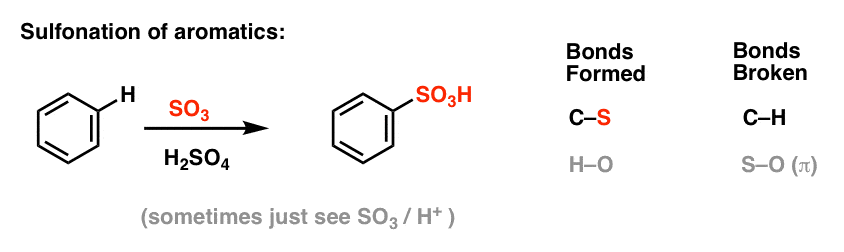
Sulfonering (ersättningen av H med en SO3H-grupp) visar sig också vara en användbar reaktion. Den kan utföras genom att tillsätta svaveltrioxid (SO3) i närvaro av svavelsyra (H2SO4) som Lewis-syra:
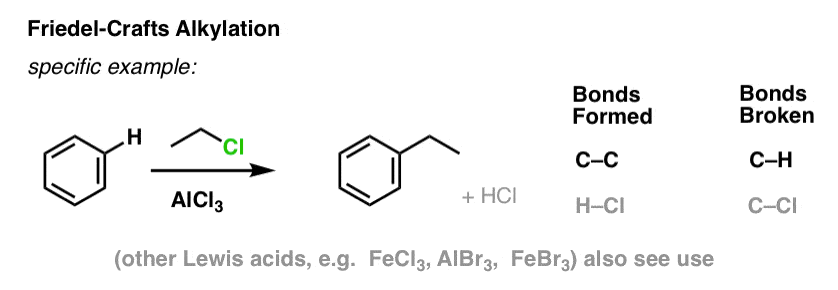
Friedel-Crafts alkylering och acylering
I de fyra reaktionerna ovan observerade vi bildandet av C-Cl, C-Br, C-N och C-S.
Det är också möjligt att bilda kol-kol-bindningar till aromatiska molekyler genom att tillsätta alkyl- eller acylhalogenider i närvaro av Lewis-syror. Dessa reaktioner är kända som Friedel-Crafts-reaktioner, efter deras uppfinnare (reaktionen går tillbaka till 1877).
I Friedel-Crafts alkylering börjar vi med en alkylhalogenid ”R-X”, till exempel CH3CH2Cl, och tillsätter sedan en Lewis-syra, till exempel AlCl3 eller FeCl3 . Precis som med Cl2 påskyndar Lewis-syran reaktionen genom att samordna sig med halogenen, försvaga C-Cl-bindningen och göra den till en bättre avgångsgrupp och därmed göra det möjligt för det bundna kolet att lättare angripas av nukleofiler. Ingen reaktion sker utan Lewis-syran.
Notera att vi här bildar C-C och bryter C-H.
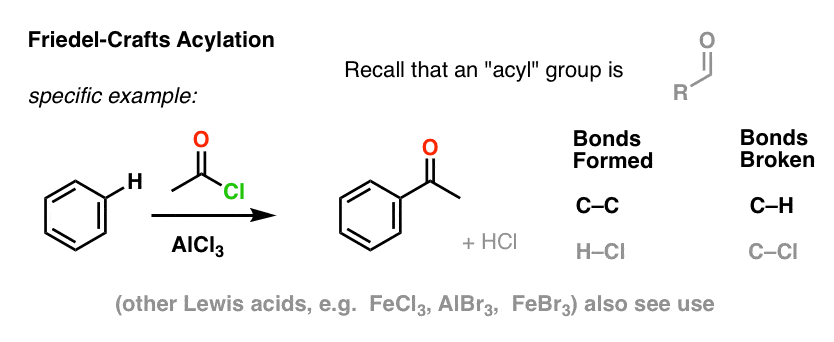
Friedel-Crafts acylering är likartad, men vi börjar med en acylhalogenid. Tillsats av vår Lewis-syra resulterar i att C-C bildas och C-H bryts.
Sammanfattning: De sex viktigaste elektrofila aromatiska substitutionsreaktionerna
Vi har visat sex viktiga elektrofila aromatiska substitutionsreaktioner (klorinering, brominering, nitrering, sulfonylering samt Friedel-Crafts alkylering och acylering) och att de alla innebär att C-H bryts och att C-E bildas (där ”E” är elektrofilen i fråga).
Men att veta vilka bindningar som bildas och bryts är bara början.
Här är några frågor som vi skulle vilja veta svaret på:
- Hur påverkar substituenter på bensen denna reaktion? Hur skulle till exempel dessa reaktioner påverkas om vi utförde dem på metylbensen? eller fenol? eller klorbensen?
- Hur påverkar elektrondonerande eller elektrondragande substituenter reaktionshastigheten?
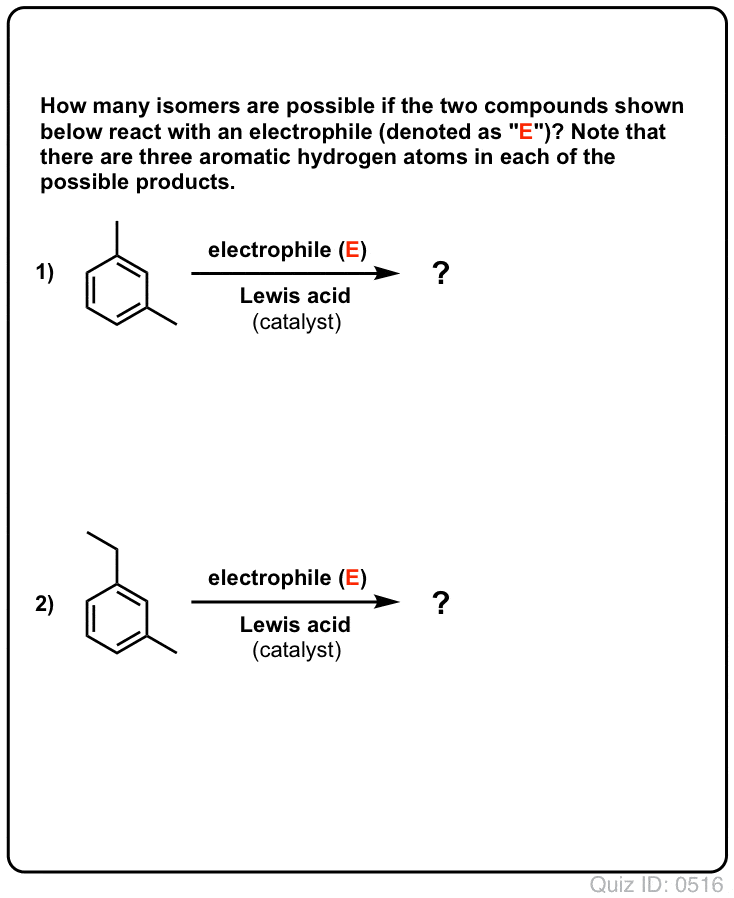
- Med bensen kan möjligen endast en monosubstituerad produkt bildas. Men vad händer om vi börjar med en monosubstituerad produkt och gör en elektrofil aromatisk substitution på den? Var hamnar substituenterna?
- Hur fungerar reaktionen? Hur förklarar vi bildandet av C-E och brytningen av C-H?
- Hur är det med andra aromatiska grupper (utöver bensen). Hur förhåller sig elektrofila aromatiska substitutionsreaktioner på pyrrol, pyridin, naftalen eller andra grupper?
I nästa inlägg kommer vi att diskutera den första frågan: effekten av elektrondonerande och elektronindragande substituenter. Vi kommer att tala om aktiverande och deaktiverande grupper.
Nästa inlägg: Tack för att du läste!
Mycket tack till Matthew Knowe för hjälpen med det här inlägget.
Testa dig själv!
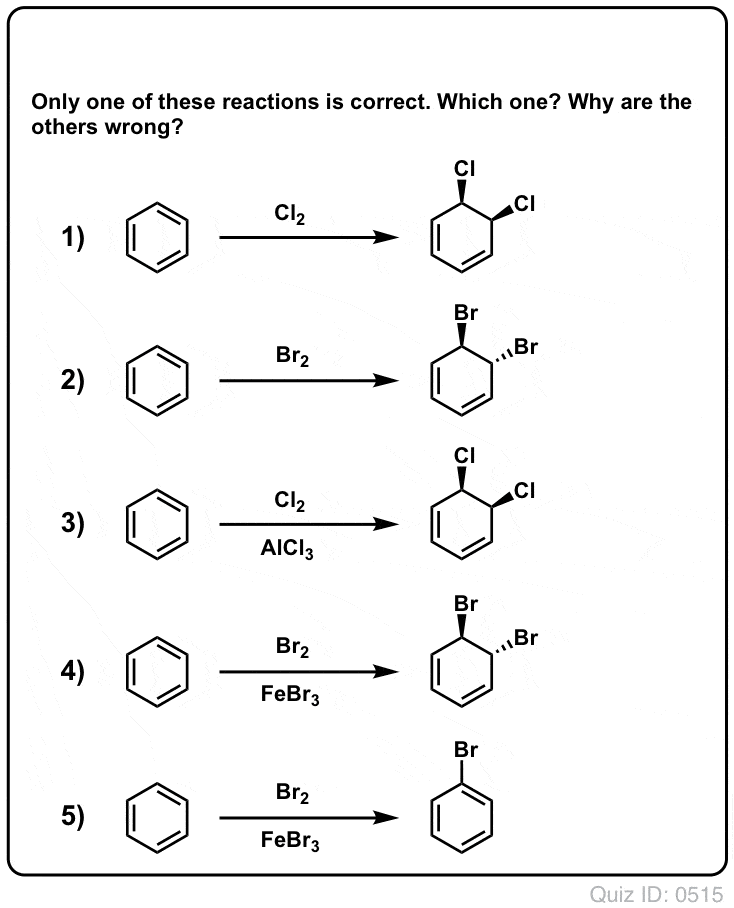 Klicka för att vända
Klicka för att vända 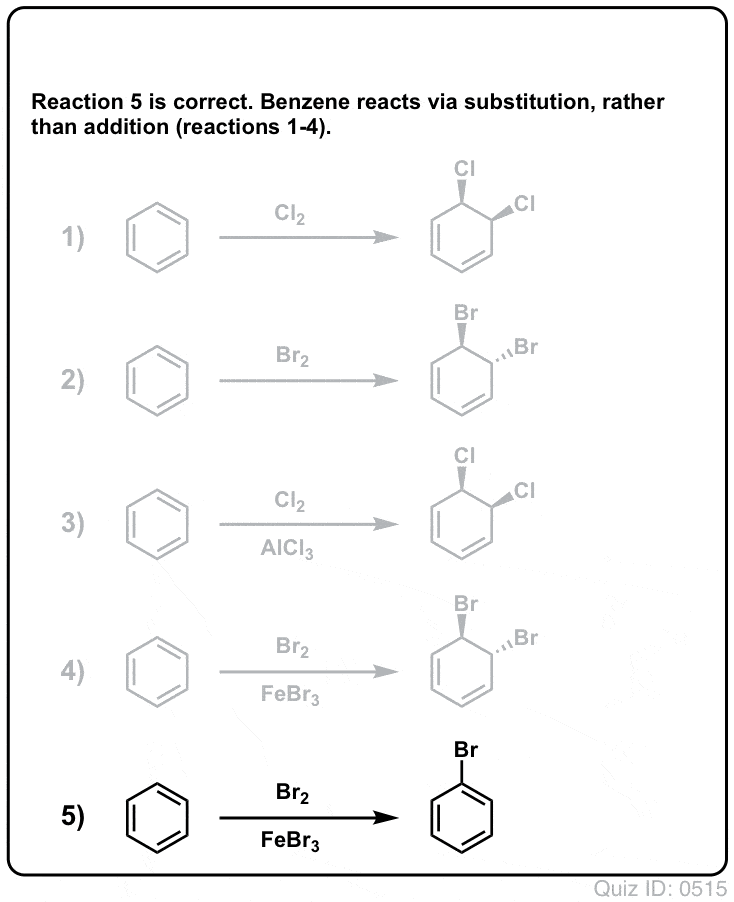
 Klicka för att vända
Klicka för att vända 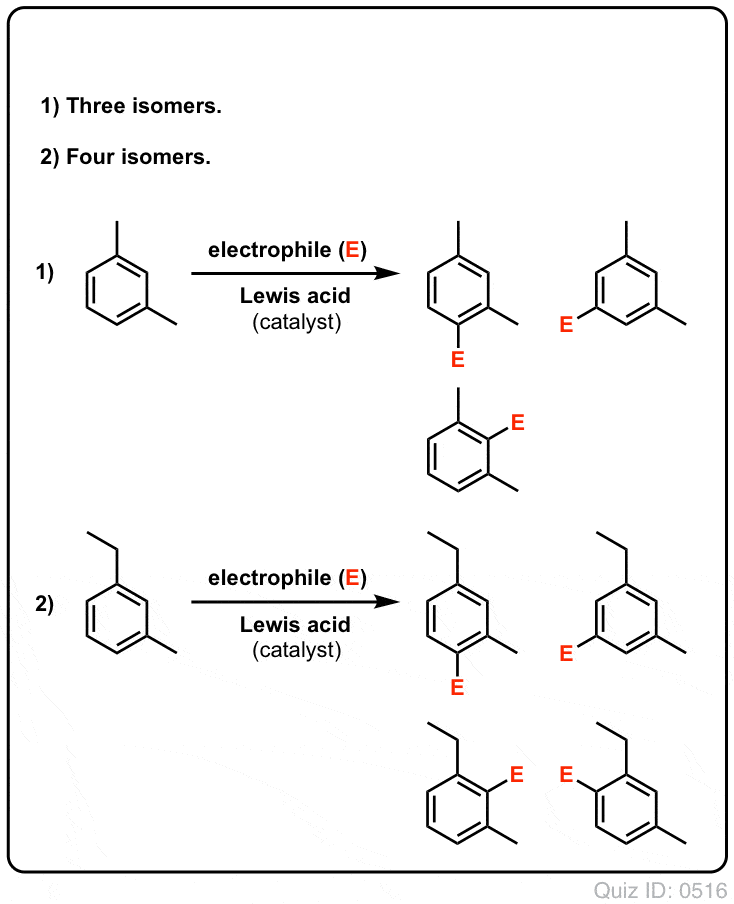
 Klicka för att vända
Klicka för att vända